Нейродегенеративные заболевания (НДЗ) — одно из активно развивающихся направлений в неврологии. В структуре неврологической патологии НДЗ занимают значительное место, являясь основной причиной деменции и различных расстройств движений [1, 3, 13]. Достижения клинической и экспериментальной медицины последних лет позволили выяснить механизмы развития этой патологии, выделить новые нозологические формы, разработать их диагностические критерии и усовершенствовать терапию [4, 9, 11]. Однако большинство практикующих врачей-неврологов испытывают затруднения в распознавании нейродегенеративных заболеваний, как правило, продолжают ошибочно трактовать их как проявления дисциркуляторной энцефалопатии и относят таких пациентов к категории бесперспективных. Между тем диагностика НДЗ на ранних этапах, использование современных лекарственных средств позволяет влиять на прогноз течения болезни, существенно улучшить качество жизни больных и даже изменить их судьбу.
Этиология и патогенез
Этиология и некоторые вопросы патогенеза НДЗ остаются неясными. В основе развития этих заболеваний лежит нарушение метаболизма и изменение конформации клеточных белков с их последующим накоплением и агрегацией в определенных группах нейронов. Эта особенность позволила отнести НДЗ к группе конформационных болезней [4, 8, 15]. Известны два белка, изменяющих структуру при НДЗ: альфа-синуклеин и тау-протеин. В соответствии с этим все НДЗ делят на два подтипа: синуклеинопатии и таупатии. Альфа-синуклеин в норме присутствует в пресинаптических терминалях головного мозга. При НДЗ данный белок накапливается и формирует внутри глиальных клеток нитевидные структуры диаметром 20—40 нм. Тау-протеин представляет собой растворимый низкомолекулярный белок, играющий важную роль в процессе роста аксона и его функционировании. При НДЗ обнаруживаются его патологические формы, образующие нити, преобладающие в телах нейронов и аксонов [14]. Причины агрегации данных белков могут как носить генетически детерминированный характер, так и быть связаны с каскадом патологических клеточных биохимических процессов: избыточного фосфорилирования, гликозилирования, активизации перекисного окисления липидов [1].
В настоящее время большинство исследователей придерживаются глутаматэргической теории нейродегенеративного процесса, предложенной в 90-е годы XX в. [5, 12]. Согласно этой теории универсальным механизмом развития всех НДЗ является эксайтотоксичность, под которой понимают повреждение и гибель нейронов в результате избыточной активации постсинаптических NMDA (N-метил-D-аспартат) рецепторов [3, 6]. В развитии каждого конкретного НДЗ играют роль определенные триггеры, к числу которых относятся недостаточность убиквитин-протеасомной системы клетки, дефекты шаперонной защиты, оксидативный стресс, апоптоз и др. [4, 5]. При НДЗ страдают преимущественно нейроны и глиальные клетки базальных ганглиев и стволовых структур, вырабатывающие ацетилхолин, дофамин, серотонин [1, 9, 14]. Недостаточность отдельных нейромедиаторов определяет клиническую картину НДЗ.
Клинические проявления
Клинические проявления НДЗ характеризуются существенным полиморфизмом за счет различных сочетаний пяти групп симптомов: экстрапирамидного, пирамидного, мозжечкового, вегетативной недостаточности и деменции [12]. В современной литературе эту группу болезней также называют «паркинсонизм плюс» в силу доминирования в клинической картине экстрапирамидных нарушений. В настоящее время используют клиническую классификацию НДЗ, согласно которой выделяют две подгруппы:
1. Спорадические НДЗ:
• Прогрессирующий надъядерный паралич (болезнь Стила —Ричардсона — Ольшевского).
• Мультисистемная атрофия.
• Деменция с тельцами Леви.
• Паркинсоническая деменция (синдром Гуам).
• Кортикобазальная дегенерация.
• Болезнь Альцгеймера.
2. Ирритативные НДЗ:
• Болезнь Гентингтона.
• Болезнь Галлервордена—Шпатца.
• Болезнь Вильсона—Коновалова.
• Болезнь Фара.
• Болезнь Бессена — Корнцвейга.
Спорадические нейродегенеративные заболевания
Прогрессирующий надъядерный паралич (ПНП, болезнь Стила — Ричардсона — Ольшевского) описан в 1964 г. одновременно J. Steele, J. Richardson и J. Olszewski [2]. Распространенность ПНП, по результатам эпидемиологических исследований, составляет 1,39 – 6,4 на 100 тыс. населения [13, 14]. При этой патологии дегенерация захватывает черную субстанцию, бледный шар, субталамическое и педункулярные ядра, таламус, ретикулярную формацию ствола, являясь по морфологическим признакам таупатией [13]. Клинические проявления ПНП чаще развиваются в возрасте 50—60 лет, в равной степени среди мужчин и женщин. Первыми нарушаются произвольные движения глаз — сначала в вертикальной, затем в горизонтальной плоскости. При этом всегда сохраняются следящие движения глаз с фиксацией объекта взором при пассивном перемещении головы (симптом кукольных глаз) [3]. Глазодвигательные нарушения сочетаются с симметричной брадикинезией (в 75% случаев) и ригидностью преимущественно в аксиальных отделах (шея, туловище). Для ПНП характерны разгибательная поза, псевдобульбарный синдром и пирамидная недостаточность [13]. Рано формируется постуральная неустойчивость в виде пропульсий, частых падений и деменция лобного типа [14]. Когнитивные нарушения при ПНП проявляются снижением способности к абстрагированию, обобщению, мышлению, обеднением речи. Течение ПНП прогрессирующее, болезнь заканчивается летально через 5—7 лет (в среднем через 87 мес) [14]. Ошибки диагностики ПНП, по данным W. Poewe [13], наблюдаются в 41% случаев.
Мультисистемная атрофия (MCA) описана J. Graham и D. Oppenheimer в 1969 г. Частота МСА составляет 1,9 – 4,4 случая на 100 тыс. населения [8]. Средний возраст начала заболевания — 60 лет, чаще страдают мужчины (соотношение 1,3:1). Отличительной морфологической чертой МСА является первичное поражение клеток глии в стриатуме, черной субстанции, голубом пятне, нижних оливах, ядрах моста, коре мозжечка, дорсальном ядре блуждающего нерва нейронов [12]. МСА относится к числу синуклеинопатий. Клинические проявления МСА характеризуется сочетанием экстрапирамидного, мозжечкового, пирамидного синдромов и прогрессирующей вегетативной недостаточности [3]. Экстрапирамидный синдром преобладает у 80% больных с МСА в форме симметричной акинезии, ригидности и постурального тремора. В 20% случаев ведущим является мозжечковый синдром в виде нарушения походки, дизартрии, динамической атаксии в конечностях. Облигатный признак МСА — вегетативная недостаточность, проявляющаяся ортостатической гипотензией, липотимиями, синкопальными состояниями. Нередко при МСА встречаются парез взора вниз, деменция подкоркового типа и миоклонии [15]. Течение заболевания прогрессирующее, продолжительность жизни после появления первых его признаков составляет 5—7 лет.
Деменция с тельцами Леви (ДТЛ) описана в начале 90-х годов прошлого века [9]. Cтрадают лица 65—70 лет, чаще мужчины.Истинная частота ДТЛ неизвестна. Морфологические признаки ДТЛ — преобладающие в коре лобной и височной долей тельца Леви, представляющие собой цитоплазматические включения, состоящие из белков альфа-синуклеина и убиквитина, а также увеличение в размерах нейронов. Характерным началом ДТЛ является триада синдромов: экстрапирамидные нарушения, деменция и галлюцинации [3, 12]. Когнитивные расстройства проявляются нарушением внимания, снижением интеллекта, потерей способности к обобщению, абстрагированию и умозаключению; отмечается инертность психических процессов [9]. Cтрадает регуляция произвольной деятельности, что подразумевает ряд последовательных актов: определение цели, построение программы и контроль за ее выполнением. Экстрапирамидный синдром при ДТЛ не имеет асимметрии, в отличие от болезни Паркинсона, и проявляется изолированной акинезией, а также выраженной постуральной неустойчивостью. Для ДТЛ характерны зрительные галлюцинации, которые четко очерчены по цвету, форме, размерам, действию и объему. Типичное проявление ДТЛ — полное исчезновение галлюцинаций при попытке взаимодействия больного с вымышленным объектом [9]. При ДТЛ нередко встречается ортостатическая гипотензия, которая проявляется липотимиями или обмороками при изменении положения тела. Для этого заболевания характерны колебания выраженности клинических симптомов в течение дня. ДТЛ отличается неуклонным прогрессированием. Спустя 2 – 3 года присоединяются тазовые нарушения в виде недержания мочи [12]. Cредняя продолжительность жизни таких больных с момента проявления первых признаков болезни — 5 лет [9].
Паркинсоническая деменция (болезнь Гуам) впервые описана у жителей островов Гуам в Тихоокеанском бассейне и относится к таупатиям. Страдают преимущественно мужчины 50 – 60 лет. При этом заболевании в клинической картине отмечаются когнитивные нарушения, синдромы паркинсонизма и бокового амиотрофического склероза. Когнитивные нарушения носят характер деменции подкоркового типа. Паркинсонизм проявляется акинезией и ригидностью преимущественно нижней части тела. Для синдрома амиотрофического синдрома характерны смешанный парез и фасцикуляции мышц верхнего плечевого пояса. Течение болезни прогрессирующее, смерть наступает через 3—5 лет [13].
Кортикобазальная дегенерация (КБД) описана в конце 90-х годов ХХ в. Встречается с частотой 0,45 на 100 тыс. населения, страдают лица 60—70 лет с одинаковой частотой у женщин и мужчин [14]. Патоморфологически при КБД поражаются нигростриарная система, таламус, субталамическое, красное и зубчатое ядра, лобная и теменная области коры. КБД относится к подтипу синуклеинопатий. Клинически заболевание проявляется асимметричной брадикинезией и ригидностью (возможно поражение противоположных конечностей (правая рука и левая нога)). Паркинсонизм часто ассоциируется с другими двигательными нарушениями (дистония, миоклонус). Для КБД характерно сочетание экстрапирамидных нарушений с апраксией, «феноменом чужой руки», чувствительными расстройствами кортикального типа, депрессией или апатией [12]. Течение заболевания прогрессирующее, смерть наступает спустя 4—8 лет.
Болезнь Альцгеймера (БА) описана в 1907 г. A. Alzheimer и в настоящее время представляет самую частую причину (до 80%) деменций в пожилом и старческом возрасте. В экономически развитых странах частота БА в возрасте до 60 лет составляет 1%, а после 60 лет удваивается через каждые 5 лет, достигая 32% в возрасте 85 лет, преобладая у женщин [1]. Патоморфологически БА принадлежит к числу таупатий и проявляется амилоидной ангиопатией, формированием сенильных бляшек [1]. Клинические проявления заболевания описаны не только в медицинской, но и в художественной литературе. Яркое описание клинических симптомов болезни встречается в рассказе И. Шоу «Солнечные берега реки Леты».
Клинические проявления БА условно разделяют на три стадии.
I стадия (начальная) проявляется изолированным ухудшением оперативной памяти или памяти на текущие события, имена, цены, названия предметов и пр. Отмечается сужение круга интересов, замедление мышления, безынициативность, рассеянность, невнимательность. Особенность данной стадии — отсутствие жалоб на ухудшение памяти вследствие нарушенной адекватной самооценки. В 50% всех случаев наблюдается сниженное настроение (депрессии) или эмоциональная неустойчивость. Бытовые и профессиональные навыки на этой стадии заболевания чаще сохранены.
II стадия (развитая) проявляется продолжающимся ухудшением кратковременной памяти, что приводит к трудностям в бытовой и производственной деятельности вследствие присоединения следующих нарушений:
• речь становится бедной, возникают сложности в подборе отдельных слов;
• нарушение целенаправленной деятельности (праксиса) заключается в трудностях выбора и надевания одежды, совершения гигиенических процедур (чистка зубов, бритье), обращения с корреспонденцией, использования домашнего оборудования; пропадает интерес к хобби; затрудняется ориентировка в незнакомой обстановке; теряется способность к вождению автотранспорта;
• нарушения оптико-пространственной деятельности: становится невозможным нарисовать любой элементарный предмет (куб, столб, циферблат часов);
• расстройство мышления (невозможность обобщения нескольких слов, интерпретации пословиц, поговорок);
• нарушение произвольного внимания и счета;
• аффективные расстройства (бред, особенно бред ревности, галлюцинации, тревога, страх).
III стадия (финальная) наступает спустя 5—10 лет от начала болезни, когда становятся невозможны любые формы мыслительной деятельности, теряется способность к самообслуживанию, речь сохраняется на уровне словесных эмболов. На этой стадии возможно присоединение потери в весе, повышение мышечного тонуса в конечностях, расстройство ходьбы, эпилептические припадки.
Ирритативные нейродегенеративные заболевания
Болезнь Гентингтона описана G. Huntington, встречается с частотой 4—10 на 100 тыс. населения в широком возрастном диапазоне (от 15 до 80 лет) [14]. Наследуется по аутосомно-доминантному типу с высокой пенетрантностью (до 50%). В результате мутаций в гене IT 15 образуется патологический белок гентингтин, который накапливается в нейронах полосатого тела. Заболевание чаще начинается в возрасте 35 — 45 лет, преобладая у мужчин. В 10% случаев возможны ранние формы (до 20 лет). Первыми симптомами болезни являются эмоциональные нарушения, депрессия либо агрессивность. Одновременно или спустя несколько лет присоединяются характерные хореические движения мышц лица (поднятие бровей), неконтролируемые движения пальцев или туловища, усиливающиеся при ходьбе и умственной нагрузке. В развитой стадии болезни появляется хореоатетоз языка, плечевого и тазового пояса, что обусловливает формирование своеобразной «танцующей» походки. Хотя при этом заболевании хорея преобладает в 90% случаев, однако при ранних формах могут наблюдаться брадикинезия, ригидность, дисфагия, мышечные дистонии и глазодвигательные нарушения [7]. У всех больных развиваются прогрессирующие когнитивные нарушения, приводящие к деменции. Средняя продолжительность жизни после появления первых признаков заболевания составляет 15—20 лет.
Болезнь Галлервордена – Шпатца (БГШ) описана в 1922 г. J. Hallervorden и H. Spatz [7]. Истинная распространенность неизвестна. Передача происходит по аутосомно-рецессивному типу. Патоморфологически при БГШ поражаются базальные ганглии за счет накопления в нейронах железа или железосодержащего фермента, вступающих в соединение с альфа-синуклеином. Заболевание может проявиться в любом возрасте. Выделяют три основные формы: 1) ранняя детская (с началом до 10 лет); 2) ювенильная (10 – 18 лет); 3) взрослая. В любом возрасте заболевание начинается с появления дистонии нижних конечностей с последующей генерализацией и вовлечением других мышечных групп (оромандибулярная, ларингеальная, туловищная, цервикальная). У половины больных БГШ в последующем развиваются гипокинезия, ригидность и постуральная неустойчивость в виде про-, ретро-, или латеропульсии, внезапных падений при ходьбе, положительная проба Тавенарда. У части пациентов возможно появление хореиформных гиперкинезов, тремора, пирамидной недостаточности, когнитивных нарушений и пигментной дегенерации сетчатки. Течение БГШ медленно прогрессирующее. При раннем начале продолжительность жизни таких пациентов составляет 10 — 15 лет, у взрослых – 15 – 40 лет [5].
Болезнь Вильсона – Коновалова (гепатоцеребральная дегенерация, ГЦД) описана A. Wilson в 1912 г. и дополнена Н.В. Коноваловым, встречается с частотой 1—2 случая на 100 тыс. населения [7]. Установлено развитие ГЦД вследствие множественных (более 100) мутаций гена ГЦД на 13 хромосоме, кодирующего синтез медь-транспортной АТФазы. Из-за генетического дефекта выведения медь в больших концентрациях аккумулируется в печени, мозге, почках, роговице, радужной оболочке глаза, образуя кольца Кайзера — Флейшера. Заболевание наследуется по аутосомно-рецессивному типу. Частота семейных случаев достигает 61% [7]. Различают три генотипических варианта ГЦД: 1) славянская (поздняя, в 20—35 лет) характеризуется неврологической симптоматикой и незначительным поражением печени; 2) западная (ювенильная, в 10—16 лет) отличается первичным поражением печени и затем появлением неврологической симптоматики; 3) атипичная (проявляется только снижением уровня церулоплазмина без клинических признаков заболевания). Неврологические проявления ГЦД характеризуются значительным клиническим полиморфизмом. Выделяют пять основных клинических форм заболевания: абдоминальная, ригидно-аритмо-гиперкинетическая, дрожательно-ригидная, дрожательная и экстрапирамидно-корковая формы [8]. Чаще встречается дрожательно-ригидная форма болезни.
Болезнь Фара (БФ) описана T. Fahr в 1930 г. [7]. Характерный рентгенологический признак БФ — массивная кальцификация подкорковых ганглиев (чаще бледного шара) и колена внутренней капсулы. БФ встречается крайне редко. По нашим данным, рентгенологические признаки БФ отмечаются в 0,04% случаев проведения КТ головного мозга, клинические проявления заболевания отмечались только в 9 % из них [8]. Главным патогенетическим механизмом развития заболевания является нарушение фосфор-кальциевого метаболизма. Его основной причиной считается первичный (аутоиммунный) или после-операционный эндокринный аденоматоз щитовидной либо паращитовидной железы, а также хронический респираторный алкалоз, приводящий к гиперкальциемии, гипонатриемии. Неврологические симптомы заболевания включают различные экстрапирамидные нарушения (ригидность, тремор, гиперкинезы), преходящие или стойкие пирамидные знаки, эпилептические приступы, деменцию. В клинической картине БФ нередко отмечаются проявления гипер- или гипопаратиреоза в форме фокальных судорог, тетанических спазмов, болей в дистальных отделах конечностей, положительные симптомы Хвостека и Труссо [8].
Нейроакантоцитоз (болезнь Бессена – Корнцвейга) описана в 1950 г. F. Bassen и A. Kornzweig. Встречается в любом возрасте в виде сочетания экстрапирамидного синдрома, полиневропатии, снижения интеллекта и изменений в крови. Паркинсонизм при этой патологии проявляется брадикинезией, ригидностью мышц шеи и конечностей, затруднением в открывании рта и высовывании языка, постуральной неустойчивостью. Полиневропатия характеризуется снижением глубоких рефлексов и нарушением всех видов чувствительности дистального типа. Когнитивные нарушения обычно слабо выражены. Патогномоничными для этой патологии считаются изменение формы и размеров эритроцитов, зазубренность их краев, может повышаться уровень креатинфосфокиназы [11].
Диагностика
Диагностика нейродегенеративных заболеваний основана на сборе жалоб и анамнеза заболевания со слов больного и (или) его родственников, общесоматического, неврологического, нейропсихологического и нейровизуализационного обследований. Достоверная информация о появлении признаков дезадаптации пациента в бытовой и (или) производственной деятельности нередко играет ведущую диагностическую роль [1]. У всех пациентов с подозрением на НДЗ наряду с рутинным соматическим и неврологическим обследованием обязательно выполнение скрининговых нейропсихологических тестов, таких как Mini Mental State Examination и тeст рисования часов, которые позволяют объективизировать степень когнитивных нарушений и различить деменцию лобного и подкоркового типов [2]. Минимальный уровень лабораторных исследований с целью дифференциальной диагностики НДЗ обычно включает общий и биохимический анализы крови (мочевина, холестерин, креатинин, билирубин, трансаминазы, фолиевая кислота, электролиты, церулоплазмин и медь), гормоны щитовидной железы, серологические и иммуноферментные исследования на сифилис и ВИЧ-инфекцию. В алгоритм обследования больных с НДЗ входят исследования глазного дня и КТ (МРТ) головного мозга. На глазном дне специфическим признаком (в 70% случаев) гепатоцеребральной дегенерации является обнаружение колец Кайзера — Флейшера [8], при болезни Галлервордена—Шпатца — атрофия зрительных нервов. Общим КТ (МРТ) признаком всех НДЗ с достоверно большей частотой является суммарная и (или) регионарная атрофия вещества головного мозга, в отличие от выраженного поражения белого вещества в перивентрикулярных зонах (лейкоареозис), что более характерно для дисциркуляторной энцефалопатии [1]. Известен ряд специфичных КТ (МРТ) признаков отдельных НДЗ. Одним из ранних диагностических признаков болезни Альцгеймера считается уменьшение объема гиппокампа [1]. КТ-признаком болезни Фара является массивная кальцификация базальных ганглиев. МРТ-проявление болезни Вильсона — Коновалова — симметричные участки гиперинтенсивного сигнала в Т1w-режиме в области бледного шара и черной субстанции, что обусловлено отложением меди. Патогномоничный МРТ-признак болезни Галлервордена — Шпатца — обнаружение в области базальных ганглиев обширной гиперинтенсивной зоны, окруженной ободком гипоинтенсивного сигнала (рисунок), связанного с отложением железа, что описывается в литературе как «глаз тигра» [6]. Современным методом диагностики НДЗ является позитронно-эмиссионная и спектрально-эмиссионная томография головного мозга, которая позволяет с помощью радиоактивных изотопов выявить билатеральное уменьшение кровотока в височно-теменных отделах коры, что, по мнению W. Poewe [13], высокочувствительно для болезни Альцгеймера.
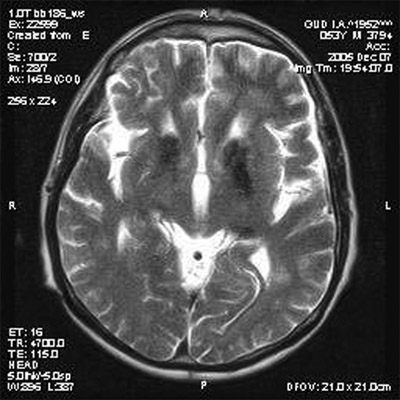
Рисунок. МРТ головного мозга больного Ш., 32 лет, с диагнозом болезнь Галлервордена – Шпатца (собственное наблюдение): в области базальных ганглиев с двух сторон регистрируется массивная зона пониженной плотности с очагом просветления в центре — «глаз тигра»
Лечение
В настоящее время наиболее распространена симптоматическая терапия нейродегенеративных заболеваний. Условиями ее успеха являются длительный курсовой характер, адекватные дозы, а также отказ от применения сомнительных по эффективности и сильнодействующих средств с выраженными побочными эффектами [8]. C целью коррекции деменции наряду с известными вазоактивными (кавинтон, сермион), комбинированными (фезам, инстенон) и ГАМК-эргическими (аминалон) средствами, эффективность которых в отношении НДЗ не доказана, в последние годы начали применяться новые группы препаратов:
• Средства, воздействующие на специфические нейромедиаторные системы мозга (реминил, арисепт, нейромидин, глиатилин). Указанные препараты являются центральными холиномиметиками, восполняют дефицит ацетилхолина в мозге за счет блокады холинэстеразы и приводят к увеличению продолжительности его действия на постсинаптические рецепторы, усиливая холинергическую передачу [1, 7]. Схожим механизмом действия обладают антиглутаматергические средства (мемантин), являющиеся неконкурентными селективными антагонистами NMDA-рецепторов. Благодаря модулированию глутаматэргической передачи улучшаются когнитивные процессы. В контролируемых исследованиях доказана эффективность препарата при всех видах деменции [4].
• Cредства с нейротрофическим действием (кортексин, церебролизин) способны повышать эффективность метаболических процессов в ткани мозга, снижать образование токсических свободных радикалов, стимулировать репаративные процессы [7].
• Cредства с нейрометаболическим действием (актовегин, глицин, семакс, луцетам, пирацетам), стимулирующие обменные процессы в головном мозге, повышают устойчивость к гипоксии, усиливают холинергическую передачу, улучшают микроциркуляцию, блокируя агрегацию тромбоцитов [3].
Определенный интерес представляет изучение зависимости эффективности пирацетама от его дозы. Диапазон доз пирацетама, применявшихся в ходе ряда многоцентровых исследований, колеблется от 1,2 до 9,6 г/сут, причем наилучшие результаты были достигнуты при приеме дозы 4,8 г/сут [7]. В Беларуси в клинической практике рекомендуется суточная доза 1200 мг пирацетама, что связано с опасностью усиления побочных эффектов, обусловленных назначением его высоких доз. По нашему опыту, хорошая переносимость высоких доз пирацетама и наличие на белорусском рынке удобных форм 800 и 1200 мг препарата в одной таблетке (луцетам) делает возможным его более широкое и эффективное использование у больных с НДЗ.
Коррекция экстрапирамидных нарушений при НДЗ возможна путем назначения леводопасодержащих препаратов (мадопар, наком), дофаминовых агонистов (мирапекс, проноран) или амантадинов (мидантан, ПК-Мерц). Эффективность препаратов леводопы при НДЗ, в отличие от болезни Паркинсона, не столь высока и, по данным W. Poewe, составляет 26% [13]. В связи с этим назначение агонистов дофаминовых рецепторов и амантадинов представляется более предпочтительным, так как указанные средства не только повышают чувствительность постсинаптических рецепторов, но и обладают антиоксидантным эффектом [9]. В случае прогрессирующей вегетативной недостаточности, не поддающейся коррекции немедикаментозными способами, эффективны кортинефф или гутрон, назначаемые в утренние часы [8]. При повышении мышечного тонуса пирамидного типа назначают баклофен, возможно его интратекальное введение с помощью специальной помпы [14]. Психотические нарушения корригируются назначением атипичных нейролептиков (клозапид). Коррекция тремора, мышечных дистоний или миоклоний возможна с помощью клоназепама, при неэффективности повторно можно ввести диспорт в заинтересованные мышцы [15]. При тяжелых формах дистонии прибегают к нейрохирургическому вмешательству — электрической стимуляции бледного шара, которая, по данным B. Biolsy et al., эффективна у 42% больных [10]. При гепатоцеребральной дегенерации обоснован пожизненный прием купренила в сочетании с препаратами цинка [8].
Перспективные направления в терапии НДЗ [8]:
• стабилизация нативной конформации клеточных белков;
• предупреждение образования агрегатов и сборки нейрофибрилл. В настоящее время уже создано несколько эффективных ингибиторов агрегации, однако в эксперименте они проявили выраженное токсическое действие. Изучается подобная способность препаратов магния [5];
• растворение образовавшихся агрегатов, но пока такая физиологическая функция не разработана;
• воздействие на вторичные цитотоксические процессы (митохондриальная дисфункция, оксидантный стресс, гиперпродукция оксида азота, активация микроглии, механизмы апоптоза);
• вакцинация при помощи введения специфических антител, однако клинические испытания прекращены из-за побочных эффектов [5].
Таким образом, нейродегенеративные заболевания представляют собой разнородную группу со сложным и до конца не ясным патогенезом. Их своевременная диагностика на ранних этапах, назначение новейших лекарственных средств способны лишь замедлить темп развития патологического процесса и улучшить качество жизни пациентов. Достижения экспериментальной и клинической медицины позволяют надеяться, что со временем появятся методы патогенетической терапии НДЗ, которые изменят пессимистическое отношение врачей к этим больным.
Литература
1. Гаврилова С.И. Практическое руководство по диагностике и лечению болезни Альцгеймера. — М., 2002.
2. Гусев Е.И., Бурд Г.С., Никифоров А.С. Неврологические синдромы, симптомокомплексы и болезни. — М.: Медицина, 1999.
3. Дамулин И.В. Некоторые аспекты дифференциальной диагностики и терапии деменций. — М., 2004.
4. Иллариошкин С.Н., Клюшников С.А., Брылев Л.В. и др. // Неврол. журнал.— 2006.— N 5. — С. 47 – 53.
5. Калвиньш И.Я. М-лы международной конференции неврологов и семейных врачей. — Рига, 2006.
6. Левин О.С., Юнищенко Н.А., Амосова Н.А. и др. // Неврол. журнал. — 2004.— N 2. — С. 36 – 46.
7. Левин О.С. Основные лекарственные средства, применяемые в неврологии.— М.: Медпресс-информ, 2006.
8. Пономарев В.В. Редкие неврологические синдромы и болезни. — СПб.: Фолиант, 2005.
9. Яхно Н.Н., Преображенская И.С.N 6. — С. 4 — 11. // Неврол. журнал. — 2003.—
10. Biolsi B., Cif L., Robles S.G. // Eur. J. Neur. — 2006. — V. 13 (Suppl. 2). — P. 20—21.
11. Bostantjonoulou S. Teaching Course Movement Disorders. — Athens, 2005.
12. Kaufmann H., Biaggoni I. Teaching Course Movement Disorders. —Athens, 2005.
13. Poewe W. Teaching Course Movement Disorders. —Athens, 2005.
14. Quinn N. Teaching Course Movement Disorders. —Copenhagen, 2000.
15. Wenning G. Teaching Course Movement Disorders. — Copenhagen, 2000.
Медицинские новости. - 2007. - №5. - С. 23—28.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.