В последние годы заметно возрос интерес к проблеме возрастных гормональных изменений в репродуктивной системе мужчины. Увеличение продолжительности жизни, особенно в развитых странах, и, следовательно, увеличение процента пожилых людей в структуре населения требуют внедрения медицинских технологий, способствующих повышению качества их жизни, поскольку на «пожилой» возраст теперь приходится треть жизни [2].
Со старением населения ассоциирована еще одна очень серьезная медицинская проблема — сахарный диабет 2-го типа (СД 2). По данным экспертов ВОЗ, в 1989 г. во всем мире насчитывалось 98,9 млн больных, страдающих СД 2, в 2000 г. — 157,3 млн. В настоящее время эта цифра приблизилась к 180 млн [1]. В 2010 г., согласно прогнозам, на Земле будут жить около 215 млн человек с СД 2-го типа.
Характерными особенностями СД 2 являются его манифестация в возрасте после 40 лет и ассоциация с избыточной массой тела и другими составляющими метаболического синдрома. Поскольку у мужчин по мере старения меняется функциональная активность всей эндокринной системы, представляет интерес изучение синдрома возрастного дефицита андрогенов при СД 2 [15].
Исследования, направленные на поиск возможных механизмов взаимосвязи нарушений углеводного обмена и дефицита андрогенов, начали активно проводиться в последние 10-15 лет. Так, уже в 90-х годах ХХ в. появились работы, указывающие на возможное участие инсулина в многофакторной системе регуляции секреции андрогенов, с одной стороны, и на влияние андрогенов на действие инсулина — с другой [14].
В ряде исследований установлено, что при гипогонадизме у мужчин имеют место инсулинорезистентность (ИР) и гиперинсулинемия, которые являются патогенетическими составляющими СД 2. Так, в исследовании TELECOM у 1292 мужчин была обнаружена четкая отрицательная корреляционная связь между уровнями тестостерона и инсулина, сохранявшаяся и после внесения поправок на вес, возраст, наличие ожирения, уровень гликемии, потребление алкоголя и курения [30]. Аналогичные результаты получены в проспективном исследовании Massachusets Male Aging Study, посвященном изучению старения мужчин. Среди прочих параметров оценивались уровень тестостерона и риск развития сахарного диабета у мужчин 40—70 лет с 10-летним интервалом [31]. Отмечено, что низкие уровни тестостерона и глобулина, связывающего половые стероиды (ГСПС), являются предикторами развития ИР и, следовательно, СД 2. Риск развития СД 2 составлял 1,58 при снижении уровня свободного тестостерона на 1 стандартное отклонение (4 нг/дл) и 1,89 — при снижении ГСПС на 1 стандартное отклонение (16 нмоль/л) [31]. Наличие связи между риском СД 2, ИР и снижением уровня андрогенной насыщенности подтверждено и в исследовании MRFIT (Multiple Risk Factor Intervention Trial) [23].
В исследовании CALDIA (CALedonia DIAbetes Mellitus) также показано, что у мужчин СД 2 ассоциирован с более низким уровнем тестостерона. Этот уровень имел отрицательную связь с индексом массы тела (ИМТ) и концентрацией инсулина натощак [12]. При исследовании 985 мужчин в возрасте от 40 до 79 лет 110 из них с СД 2 имели существенно более низкий уровень общего тестостерона и ГСПС, нежели мужчины без этого заболевания, с учетом поправки на возраст и ИМТ. В то же время в группах сравнения различий в уровнях эстрадиола и андро-стендиона не выявлено [12].
Однако в ряде публикаций приведенные закономерности подвергаются сомнению. Так, в исследованиях R. Y. Chen et al., несмотря на более низкий уровень общего тестостерона у мужчин с метаболическим синдромом по сравнению с группой здоровых и наличие обратной корреляционной связи между его концентрацией и окружностью талии, а также уровнем холе-стерина липопротеидов низкой плотности (ЛПНП), взаимосвязь между низким уровнем тестостерона и риском развития СД 2 не установлена [10].
При анализе данных литературы, свидетельствующих о наличии связи между андрогенным дефицитом и СД 2, закономерно возникает вопрос: что же первично — снижение уровня тестостерона (обусловленное возрастными изменениями), которое вносит определенный вклад в развитие ИР и СД 2, или наличие СД 2, ведущего к снижению секреции тестостерона? Ответ на данный вопрос очень важен, поскольку он предполагает определение патогенетических подходов к лечению как СД 2, так и возрастного андрогенного дефицита. По мнению G. Tibblin et al., снижение уровня тестостерона является не только фактором риска возникновения ожирения, но и независимым фактором риска возникновения СД 2, поскольку дефицит тестостерона ведет к увеличению количества абдоминальной жировой клетчатки и развитию ИР [34].
Позитивное влияние экзогенно вводимых андрогенов у мужчин с СД 2 и возрастным андрогенным дефицитом было продемонстрировано M.A. Boyanov et al. В частности, отмечено, что применение андрогенов ведет к улучшению метаболического контроля сахарного диабета (снижается уровень гликированного гемоглобина (HbA1c), вес, уменьшается соотношение объем талии/объем бедер, которое отражает уменьшение количества висцеральной жировой ткани) [7].
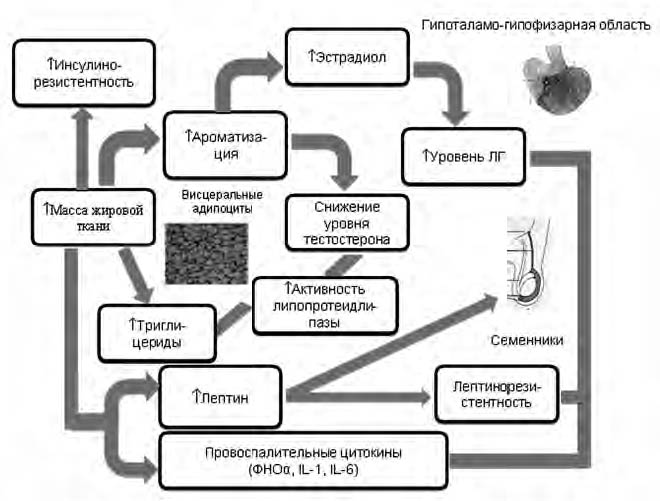
Рис.1. Гипогонадально-жировой адипоцитокиновый цикл [15]
На рис. 1 представлен гипогонадально-жировой адипоцитокиновый цикл, определяющий взаимоотношения между андрогенной функцией и висцеральным ожирением. Видно, что абдоминальное ожирение ведет к повышению активности ароматазы, присутствующей в жировой ткани и конвертирующей тестостерон в эстрогены. В то же время снижение уровня тестостерона увеличивает активность липопротеидлипазы и уровень триглицеридов, что еще больше усугубляет ожирение и приводит к ИР. Это, в свою очередь, вызывает дальнейший дефицит андрогенов и накопление висцерального жира. Кроме того, уровни тестостерона снижаются в результате лептинрезистентности на гипоталамо-гипофизарном уровне и ингибирующего влияния лептина на тестикулы. Провоспалительные адипоцитокины типа фактора некроза опухолей-α (ФНО-α) и интерлейкина-6 (ИЛ-6) могут также ингибировать гипофизарную область, приводя к снижению уровня тестостерона [11, 16].
Адипоциты висцеральной жировой ткани секретируют свободные жирные кислоты, попадающие в воротную вену печени. Высокие концентрации свободных жирных кислот подавляют поглощение инсулина печенью, что приводит к гиперинсулинемии и относительной ИР в сочетании с гипергликемией и гипертриглицеридемией. Развивающаяся благодаря избыточному накоплению висцеральной ткани ИР является связующим звеном между ожирением, нарушением толерантности к глюкозе, артериальной гипертензией и дислипидемией [29].
A.H. Kissebah, P. Bjorntorp показали, что при одинаковых показателях ИМТ абдоминальное ожирение сопровождается более высоким риском развития сопутствующих заболеваний, нежели периферическое. Ввиду того, что абдоминальное ожирение более часто регистрируется у лиц мужского пола, выраженность осложнений ожирения у них более существенная [3, 18]. По данным некоторых исследователей, осложнения от ожирения в большей мере зависят от распределения жира, нежели от степени ожирения [4, 13].
Как известно, в жировой ткани представлены рецепторы к инсулину, глюкагону, соматотропному и тиреотропному гормонам, глюкокортикостероидам, тиреоидным гормонам, а также к половым гормонам — андрогенам, эстрогенам и прогестерону [17]. Ароматаза избыточной жировой ткани в повышенных количествах превращает андрогены (тестостерон и андростендион) в эстрогены (эстрон и эстрадиол соответственно). Последние подавляют секрецию гонадотропин рилизинг-гормона и ЛГ, что проявляется снижением уровня тестостерона [17].
Доказано, что дефицит тестостерона, определяющий наличие гипогонадизма, — мощный стимулирующий фактор роста висцеральных адипоцитов. Тестостерон, как эндогенный, так и экзогенный, снижает количество висцерального жира, воздействуя на специфичные андроген-рецепторы в адипоцитах и стимулируя аденилатциклазу, протеинкиназу А и гормонспецифическую липазу; в результате потенцируется липолиз и снижается количество жира в адипоцитах [5].
Патогенетические механизмы определения и подтверждения взаимосвязи ИР, висцерального ожирения и андрогенного дефицита включают анализ вариабельности факторов воспаления, продуцируемых адипоцитами, — С-реактивного белка (СРБ), некоторых цитокинов (ФНО-α), интерлейкинов (ИЛ-1, ИЛ-6 и др.).
В настоящее время не вызывает сомнений наличие положительной корреляции между массой тела, количеством висцерального жира, ИР, с одной стороны, и ФНО-α, ИЛ-6, лептина, ингибитора активатора плазминогена-1 и т.д. — с другой [6, 21, 24, 25, 28].
Отмечена положительная корреляция между экспрессией ФНО-α и индексом массы тела, систолическим артериальным давлением, а также снижением продукции ФНО-α и его концентрации в крови при уменьшении массы тела [6]. V. Mohamed-Ali et al. высказано предположение, что цитокин ФНО-α оказывает ауто- и паракринное действие и имеет наибольшее значение для развития ИР жировой ткани [21]. Кроме того, ФНО-α может способствовать ИР путем стимуляции липолиза в адипоцитах [25, 37] и повышению уровня ингибитора активатора плазминогена-1, продукции лептина, ИЛ-6 [24, 28].
L. Roytblat et al. [26] показали, что пропорционально нарастанию массы жировой ткани концентрация ИЛ-6 в крови больных с СД 2 увеличивается на 30—40% по сравнению со здоровыми лицами и коррелирует с уровнем ИР. В другом исследовании K. Stenlof et al. продемонстрировали отрицательную корреляцию между содержанием ИЛ-6 в цереброспинальной жидкости и степенью ожирения [32]. Уменьшение массы жировой ткани на 30% в исследовании J.M. Bruun et al. привело к снижению на 25—30% концентрации провоспалительных маркеров (ИЛ-6, ФНО-α), а также к повышению чувствительности тканей к инсулину [9]. Согласно экспериментальным данным (Araneo et al., 1991; Gornstein et al., 1999; Hatakeyama et al., 2002 и др.), уровни маркеров воспаления (ФНО-α, ИЛ-6, ИЛ-1, интерферон-α) при введении андрогенов (тестостерон, дегидротестостерон и дегидроэпиандростерон) снижаются. Приведенные результаты были воспроизведены в клиническом исследовании M. Maggio et al., которые при обследовании 467 мужчин старше 65 лет выявили статистически значимую отрицательную корреляцию между уровнем ИЛ-6 и содержанием как общего, так и свободного тестостерона.
В то же время в исследованиях C.J. Malkin et al. продемонстрировано положительное влияние заместительной терапии эфирами тестостерона на маркеры воспаления: уровни провоспалительных цитокинов (ФНО- α и ИЛ-1 α) на фоне лечения достоверно снижались, а концентрация противовоспалительного маркера ИЛ-10, напротив, повышалась (рис. 2) [19, 20].
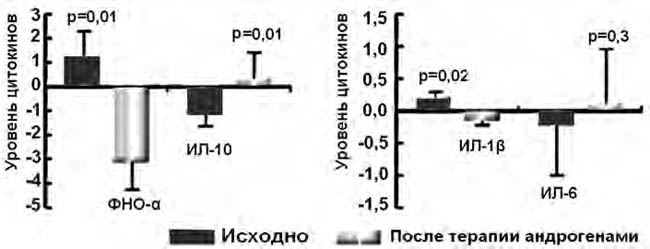
Рис. 2. Изменение уровней цитокинов на фоне терапии андрогенами [20]
Таким образом, гипоандрогения, проявляющаяся низкой концентрацией свободного тестостерона, приводит к повышению уровня маркеров воспаления и, очевидно, может способствовать развитию и прогрессированию ожирения и акселерации атеросклероза. Исходя из вышесказанного логично предположить, что нормализация уровня тестостерона будет способствовать регрессу метаболических нарушений не только за счет усиления липолиза, повышения физической активности, возрастания чувствительности тканей к инсулину, но и посредством подавления маркеров воспаления.
Продукция кортикостероидов и эстрадиола у мужчин остается постоянной на протяжении всей жизни. Напротив, продукция лептина — относительно недавно описанного гормона, вырабатываемого адипоцитами, нарушается при гипогонадизме, что объясняет, в частности, некоторые изменения в перераспределении жира, наблюдаемые у пациентов с гипогонадизмом [8].
Комплексное влияние андрогенов на метаболизм адипоцитов осуществляется по нескольким направлениям: прямое воздействие на жировую ткань, повышение ее чувствительности к инсулину, стимуляция липолиза в адипоцитах путем повышения экспрессии α-адренорецепторов, аденилатциклазы, протеинкиназы А, гормонзависимой липазы и подавления воспалительного ответа [5].
В ряде исследований анализировалась выраженность дислипидемии при использовании андрогенов у лиц с метаболическим синдромом. Большинство авторов отмечают значительное снижение уровня общего холестерина на фоне терапии андрогенами [22, 33, 36]. Ни в одном из исследований не наблюдался рост уровня холестерина липопротеидов низкой плотности (Х ЛПНП), т.е. не констатировалось негативное влияние тестостерона на липидный спектр; более того, в некоторых работах указывалось на значительное снижение уровня Х ЛПНП в крови [33, 35]. B.S. Uyanik et al. отметили прирост холестерина липопротеидов высокой плотности в ответ на терапию тестостероном у пациентов с гипогонадизмом и изначально нормальным уровнем тестостерона в крови.
Итак, ИР, являющаяся основным фактором в патогенезе метаболического синдрома, включая висцеральное ожирение и СД 2, тесно ассоциирована с нарушением (снижением) продукции тестостерона, что оправдывает включение заместительной терапии андрогенами (при наличии андрогенного дефицита) в комплекс патогенетического лечения наряду с рекомендациями по изменению образа жизни, гипогликемизирующими, гипотензивными и гиполипидемическими средствами.
Исследования последних лет убедительно продемонстрировали, что использование препаратов тестостерона приводит к улучшению некоторых параметров метаболического синдрома. В частности, применение оксандролона по 20 мг в день в течение 12 нед. способствовало значительному уменьшению количества абдоминальной жировой ткани и увеличению чувствительности к инсулину [27]. По другим данным, трехмесячная заместительная терапия препаратами тестостерона улучшает чувствительность к инсулину, снижает уровни гликированного гемоглобина, гликемии натощак и общего холестерина, а также уменьшает окружность талии. Аналогичные результаты получены P.Y. Liu et al. на фоне лечения хорионическим гонадотропином.
В настоящее время для проведения заместительной терапии гипогонадизма существует достаточно широкий спектр препаратов тестостерона, включающий пероральные формы, масляные растворы для внутримышечных инъекций, трансдермальные гели и пластыри (таблица). Поскольку все препараты тестостерона имеют свои преимущества и недостатки, при выборе метода терапии необходимо руководствоваться принципами эффективности, безопасности и удобства применения.
|
|
|
|
Тестостерона ципионат (Депо-тестостерона ципионат)
Смеси эфиров тестостерона (Сустанон 250, Омнадрен 250)
Тестостерона ундеканоат (Небидо)
|
Метилтестостерон*
Тестостерона ундеканоат (Андриол)
|
Андродерм,
Тестодерм (пластырь)
|
Таблица. Зарегистрированные в Республике Беларусь препараты тестостерона, применяемые для заместительной терапии (*17α-алкилированный препарат тестостерона, обладает выраженной гепатотоксичностью)
В заключение отметим, что многочисленные исследования дают основание расценивать гипогонадизм у мужчин как один из возможных компонентов метаболического синдрома и рекомендовать заместительную терапию андрогенами в комплексном лечении этого синдрома. Активное выявление и коррекция гипогонадизма у мужчин пожилого возраста позволят предупредить развитие СД 2 и улучшить углеводный обмен при манифестной форме заболевания, а следовательно, существенно повысить качество жизни пациентов.
Литература
1. Дедов И.И., Шестакова М.В., Максимова М.А. Федеральная целевая программа «Сахарный диабет»: метод. рекомендации. — М.: МедиаСфера, 2003.
2. Дедов И.И., Калинченко С.Ю. Возрастной андрогенный дефицит у мужчин. — М., 2006.
3. Bjorntorp P. // J. Intern. Med. — 1991. — Vol. 230. — P. 195—201.
4. Bjorntrop P. // Arteriosclerosis. — 1990. — Vol. 10. — P. 493—496.
5. Bjоrntorp P. // Curr. Opin. Lipidol. — 1994. — Vol. 5. — P. 166—174.
6. Bogdanski P., Kujawska-Luczak M., Lacki J. et al. // Pol. Merkuriusz Lek. — 2003. — Vol. 15, N 88. — P. 347—349.
7. Boyanov M.A., Boneva Z., Christov V.G. // Aging Male. — 2003. — Vol. 6. — P. 1—7.
8. Bray G.A., York D.A. // J. Clin. Endocrinol. Metab. —1997. — Vol. 82. — P. 2771.
9. Bruun J.M., Verdich C., Toubro S. et al. // Eur. J. Endocrinol. —2003. — Vol. 148, N 5. — P. 535—542.
10. Сhen R.Y., Wittert G.A., Andrews G.R. // Diabetes Obes. Metab. — 2006. — Vol. 8, N 4. — P. 429—435.
11. Cohen P. G. // Med. Hypotheses. — 1999. — Vol. 52. — P. 49—51.
12. Defay R. et al. // Intern. J. Obes. — 1998. — Vol. 22. — P. 927—934.
13. Despres J.P., Moorjani S., Lupien P.J. et al. // Arteriosclerosis. — 1990. — Vol. 10. — P. 497—511.
14. Ebeling P. et al. // Clin. Endocrinol. — 1995. — Vol. 43. — P. 601—607.
15. Hugh J. T. // Eur. Endocrinol. Dis. — 2007. — Vol. 1. — P. 65—69.
16. Kapoor D., Clarke S., Stanworth R. et al. // Eur. J. Endocrinol. — 2007. — Vol. 156. — P. 595—602.
17. Kershaw E.E., Flier J.S. // J. Clin. Endocrinol. Metab. — 2004. — Vol. 83. — P. 1407—1413.
18. Kissebah A.H., Krakower G.R. // Physiol. Rev. — 1994. — Vol. 74. — P. 761—811.
19. Malkin C.J., Pugh P.J., Jones R.D. et al. // J. Endocrinol. — 2003. — Vol. 178. — P. 373—380.
20. Malkin C.J., Pugh P.J., Jones R.D. et al. // J. Clin. Endocrinol. Metab. — 2004. — Vol. 89. — P. 3313—3318.
21. Mohamed-Ali V., Pinkney J.H., Coppack S.W. // Intern. J. Obes. Relat. Metab. Disord. — 1998. — Vol. 22. — P. 1145—1158.
22. Morley J.E., Perry H.M., Kaiser F.E. et al. // J. Amer. Geriatr. Soc. — 1993. —Vol. 41. — P. 149—152.
23. Muller M., Grobbee D.E., den Tonkelaar I. et al. // J. Clin. Endocrinol. Metab. — 2005. — Vol. 90. — P. 2618—2623.
24. Plomgaard P., Keller P., Keller C. et al. // J. Appl. Physiol. — 2005. — Vol. 98, N 6. — P. 2019—2023.
25. Porter M.H., Cuthins A., Fine J.B. et al. // J. Clin. Med. — 2002. — Vol. 139. — P. 140—146.
26. Roytblat L., Rachinsky M., Fisher A. et al. // Obes. Res. — 2000. — Vol. 8, N 9. — P. 673—675.
27. Schroeder E.T., Zheng L., Ong M.D. et al. // J. Clin. Endocrinol. Metab. — 2004. — Vol. 89. — P. 4863—4872.
28. Sethi J.K., Hotamisligil G.S. // Semin. Cell Dev. Biol. — 1999. — Vol. 10, N 1. — P. 19—29.
29. Simon D., Charles M.A., Lahlou N. et al. // Diabetes Care. — 2001. — Vol. 24. — P. 2149—2151.
30.Simon D. et al. // J. Clin. Endocrinol. Metab. — 1997. — Vol. 82, N 2. — P. 682—685.
31. Stellato R.K., Feldman H.A., Hamdy O. et al. // Diabetes Care. — 2000. — Vol. 23, N 4. — P. 490—494.
32. Stenlof K., Wernstedt I., Fjallman T. et al. // J. Clin. Endocrinol. Metab. — 2003. — Vol. 88, N 9. — P. 4379—4383.
33. Tenover J.S. // J. Clin. Endocrinol. Metab. — 1992. — Vol. 75. — P. 1092—1098.
34. Tibblin G. et al. // Diabetes. — 1996. — Vol. 45, N 11. — P. 1605—1609.
35. Uyanik B.S., Ari Z., Gumus B. et al. // Jpn. Heart J. — 1997. — Vol. 38. — P. 73—82.
36. Zgliczynski S., Ossowski M., Slowinska-Srzednicka J. et al. // Atherosclerosis. — 1996. — Vol. 121. — P. 35—43.
37.Zinman B., Hanley A.J.G., Harris S.В. et al. // J. Clin. Endocrinol. Metab. —1999. — Vol. 84. — P. 2172—2178.
Медицинские новости. – 2008. – №3. – С. 14-17.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.