Мозговой инсульт – одно из наиболее частых проявлений при сахарном диабете (СД), эта патология развивается более чем у 700 000 пациентов в год. В США мозговой инсульт является 3-й причиной смерти и нетрудоспособности при СД. СД – наиболее важный фактор риска развития ишемических инсультов и тесно ассоциирован с повышением не только частоты заболеваемости, но и смертности [5].
По результатам многолетнего эпидемиологического исследования было установлено, что частота новых случаев ишемического инсульта среди лиц с сахарным диабетом достигает 62,3 на 1000 человек, в то время как в основной популяции она составляет 32,7 на 1000 человек в течение 12-летнего периода наблюдения [41, 4]. Относительный риск развития инсульта выше у лиц с СД 2-го типа в 1,8–6 раз по сравнению с лицами без СД. В исследовании MRFIT риск смерти от инсульта среди пациентов с СД был в 2,8 раза выше по сравнению с пациентами без сахарного диабета, при этом риск смерти от ишемического инсульта был выше в 3,8 раза, от субарахноидального кровоизлияния – в 1,1 раза и от внутримозгового кровоизлияния – в 1,5 раза [39]. Вместе с тем, частота случаев геморрагического инсульта не отличается от таковой в общей популяции [29, 36]. СД является фактором риска развития нарушений мозгового кровообращения независимо от наличия других факторов риска (артериальная гипертензия, гиперхолестеринемия). У большинства (72–75%) больных сахарным диабетом отмечается не тромботический характер инсульта, что превышает стандартные показатели (среди населения в целом – 60%). В развитии нетромботического инфаркта мозга при СД ведущие роли принадлежат хронической ишемии мозга и поражению симпатических вазомоторных нервов.
Существенную роль в развитии цереброваскулярных нарушений играет патология магистральных артерий головы: сонных и позвоночных артерий, которые при сахарном диабете часто поражаются атеросклерозом. Доказано, что СД и гипергликемия являются независимыми факторами риска развития системного атеросклероза с поражением сосудов различных локализаций, в том числе мозговых. При этом подавляющее большинство работ посвящены оценке атеросклеротического поражения коронарных артерий и артерий нижних конечностей, поражение которых сопровождается повышением в 2–4 раза риска смерти от инфаркта миокарда и ампутаций нижних конечностей [41]. Кроме того, для сахарного диабета характерно системное поражение сосудов микроциркуляторного русла (микроангиопатия), которое сопровождается развитием нарушений микроциркуляции в органе-мишени, т.е. мозге. Микроангиопатия мозговых сосудов усугубляет метаболические нарушения, развивающиеся при хронической ишемии мозга и повышает риск деменции [37].
Важность изучения влияния глюкозы и инсулина на толщину мышечного слоя артерий (индекс IMT) подтверждена работами, проводившимися в рамках международной программы IRAS [18].
Вторым проявлением цереброваскулярной патологии при сахарном диабете является когнитивная дисфункция и синдром деменции [10, 12]. Согласно метаанализу К.V. Allen и соавт., в 8 из 9 пролонгированных исследований (длительностью от 5 до 12 лет) подтверждена связь СД с когнитивными нарушениями и повышенным риском деменции [7]. При этом отмечается повышение риска развития болезни Альцгеймера при СД (см. рисунок).
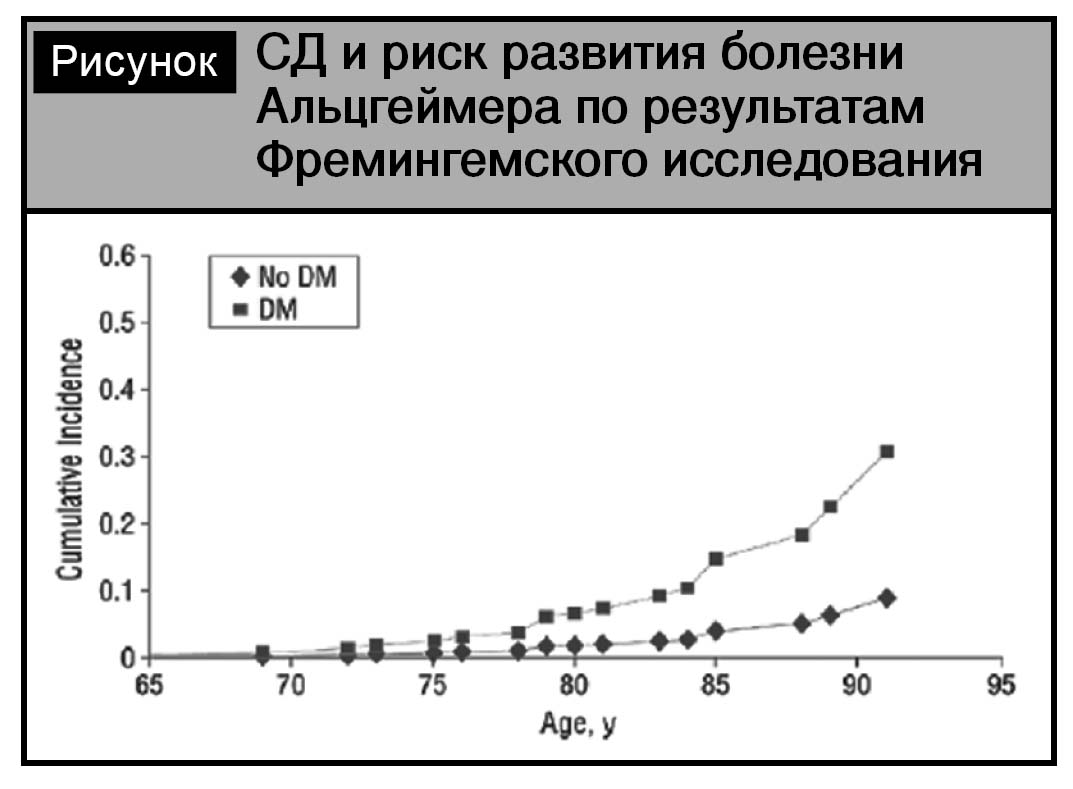
Доказано, что индекс гипогликемий нарастает с длительностью СД более 6 лет, при этом у пациентов выраженные гипогликемии ассоциированы с высоким риском деменции, а дополнительный риск деменции между лицами без гипогликемических эпизодов и с зарегистрированными эпизодами составил 2,39% в год [8]. Многочисленные публикации свидетельствуют, что при СД отмечаются снижение скорости психомоторных реакций, нарушение функции лобной доли, снижение вербальной памяти, комплексные моторные нарушения, визуальные задержки, снижение внимания и др. [13, 42].
Считается, что когнитивные и нейрональные нарушения при СД могут быть обусловлены прямым влиянием ятрогенных гипогликемий, другими метаболическими нарушениями (метаболический компонент деменции), нарушениями перфузии мозговой ткани (сосудистый компонент деменции), ассоциацией с депрессивными состояниями и другими факторами [10, 12].
Доказано, что острые и повторные гипогликемии снижают уровень гликогена в церебеллуме, коре и гипоталамусе на 50% по сравнению с нормальными значениями [19]. При этом после острой гипогликемии уровень гликогена возвращается к нормальным значениям через 6 ч, а при повторных гипогликемиях возращение уровня гликогена происходит через 24 ч, что связывают с нарушением контррегуляторных механизмов компенсации гиперинсулинемических гипогликемий [13, 43]. Есть мнение, что гипогликемия инициирует программированную гибель или повреждение нейронов за счет биоэнергетической дисфункции мозга [14, 42].
Патогенез деменции сложен, так как включает влияние различных факторов: нарушения макро- и микроциркуляции, изменения в ткани могза вследствие метаболических нарушений. Когнитивная дисфункция при СД связана с развитием глиоза и демиелинизации нервных волокон, повышением накопления миоинозитола с развитием атрофии в области гиппокампа и префронтальной зоны лобной доли и сосудистыми нарушениями в белом веществе [24].
Развивающаяся при СД артериальная гипертензия, является причиной развития гипертонической энцефалопатии, вызывающей повреждение белого вещества головного мозга (лейкоареоз), сопровождающееся разобщением коры головного мозга и базальных ганглиев, т. е. нарушением кортикостриатопаллидоталамических связей, играющих важную роль в реализации когнитивных и двигательных функций. Морфологические изменения в участках лейкоареоза включают диффузную демиелинизацию нервных волокон, пролиферацию и гипертрофию астро-глии, отек олигодендроглиоцитов, очаги неполного некроза в белом веществе с образованием мелких полостей, расширение перивентрикулярных пространств, а также формирование спонгиоформной структуры белого вещества [33].
Основными факторами, инициирующими метаболические нарушения при СД, являются хроническая гипергликемия и выраженные гипогликемические эпизоды [11]. Хроническая гипергликемия определяет активцию инсулиннезависимых путей метаболизма глюкозы, в частности – полиолового шунта с накоплением глутатиона и активацией протеинкиназы С. Одновременно отмечается повышение внутриклеточной осмолярности с нарушением работы K+/Na+ насоса; активируются реакции оксидативного стресса c накоплением свободно-радикальных продуктов и NO-зависимая вазодилатация сосудов; процессы энзиматического и неэнзиматического гликозилирования глюкозы; снижается продукция факторов роста (нейротрофилов), повышается адгезия лейкоцитов, активируются макрофаги и процессы свертывания крови. Перечисленные нарушения приводят к активации генов, определяющих повреждения нервной ткани и сопровождаются развитием глиоза, демиелинизацией и повышением накопления миоинозитола [15].
Выраженные гипогликемические эпизоды также оказывают негативное влияние на состояние мозга, так как нарушая питание и оксигенацию мозговой ткани вызывают селективные нейрональные некрозы; сопровождаясь повышением уровня кальция, оказывают токсическое внутриклеточное действие и др. [35].
Тяжесть повреждения мозга при СД определяется cтепенью и длительностью снижения мозгового кровотока и нарушениями метаболических реакций мозга [5, 13, 30]. При снижении кровотока сначала происходит торможение белкового синтеза, затем отмечается активация анаэробного гликолиза, увеличение концентрации лактата с развитием лактат-ацидоза и тканевого цитотоксического отека. Следующая стадия характеризуется снижением синтеза АТФ, дисфункцией каналов активного ионного транспорта, дестабилизацией клеточных мембран, выбросом возбуждающего нейротрансмиттера глутамата, и при уменьшении кровотока до 20% от нормы развивается аноксическая деполяризация мембран, необратимое поражение клеток. В центре ишемического очага в течение нескольких минут наступает некроз, а вокруг «ядра» инфаркта располагается ишемическая полутень, где изменения возможно обратимы. Именно зона ишемии является основной точкой приложения метаболических средств при остром или хроническом ишемическом поражении мозга. Основными механизмами по-вреждения мозга при ишемии являются глутаматергическая эксайтотоксичность и оксидантный стресс [32].
При инсульте отмечается повышение содержания глутамата в нейронах, в цереброспинальной жидкости и в крови, что вызывает приток ионов кальция (Са2+) внутрь клетки через NMDA-рецепторы (N-метил-D-аспартатного рецептора). Кроме того, глутамат участвует в образовании оксида азота (NO) и активации протеинкиназы С, протеаз, эндонуклеаз, фосфолипаз и протеинолиза, а также активации перекисного окисления липидов клеточных мембран [30, 32].
Другой важнейший патогенетический механизм апоптоза нейронов в условиях ишемии — оксидативный стресс, характеризующийся образованием избытка свободных радикалов, способных по-вреждать белки, жиры и ДНК. Оксидантный стресс развивается при нарушении соотношения между продукцией реактивных форм кислорода и антиоксидантной защитой организма (супероксиддисмутаза, каталаза, витамины Е, С). Нейроны, особенно клетки стриатума, гиппокампа и коры головного мозга, восприимчивы к оксидативному повреждению, что определяет возможность дополнительных влияний и обусловлено большим спектром липидов, участвующих в свободнорадикальных реакциях. Токсические проявления, развивающиеся в нервной ткани, результируются в развитие локального воспаления и активацию апоптоза.
Клинические проявления цереброваскулярной патологии при СД весьма разнообразны.
Лейкоареоз, характеризующий гипертонические и постгипогликемические поражения мозга, может быть бессимптомным или проявляться сочетанием когнитивных расстройств (без очаговых нарушений высших корковых функций), прогрессирующих до синдрома деменции, и различных неврологических нарушений (обмороки, судорожный, подкорковый, псевдобульбарный и мозжечковый синдромы) [1]. При этом пациенты предъявляют ряд жалоб на головные боли, головокружение, изменения настроения, чаще с депрессивными симптомами.
Частота выявления цереброваскулярной патологии определяет необходимость использования различных лекарственных средств с учетом понимания различных патогенетических подходов к достижению нейропротекторного эффекта.
Одним из патогенетических подходов для достижения этой цели является метаболическая терапия, направленная на улучшение пластичности здоровой ткани, усиление регенераторно-репаративных процессов, активацию образования полисинаптических связей, увеличение количества рецепторов. Среди препаратов из группы активаторов метаболизма используются различные сывороточные гемодериваты (CГД) – депротеинезированные дериваты из крови здоровых молочных телят, содержащие широкий спектр низкомолекулярных компонентов клеточной массы и сыворотки крови с молекулярной массой 5000 Д (гликолипиды, нуклеозиды и нуклеотиды, олигопептиды и аминокислоты) [2, 17, 21, 26].
Проведенные экспериментальные исследования подтвердили, что депротеинезированные дериваты из крови здоровых молочных телят снижают глутаматергическую цитотоксичность нейронов, что позволяет объяснить один из механизмов эффекта воздействия на мозг. Кроме того, CГД обладают выраженным антиоксидантным эффектом, ингибируя проявления активции оксидативного стресса и оказывают влияние на процессы эксайтотоксичности – избыточной активации аминокислотных рецепторов нейронов [27]. Доказано, что сывороточные гемодериваты являются мощными антиоксидантами, так как они обладают высокой супероксиддисмутазной активностью и обеспечивают снижение уровней свободных радикалов [16]. В то же время сывороточные гемодериватыстимулируют утилизацию головным мозгом глюкозы в условиях гипоксии за счет усиления ее транспорта и накопления в клетках, а также увеличивают поглощение кислорода тканями и оказывают инсулиноподобное действие, повышая толерантность организма к глюкозе.
Механизм действия сывороточных гемодериватов обеспечивает возможность оптимизации пути энергетического обеспечения, в результате чего увеличивается синтез макроэргических соединений и улучшаются метаболиче-ские процессы в пораженных тканях мозга и поддерживается белково-синтетическая функция нейронов [27].
Нейрогенные эффекты сывороточных гемодериватов используются с 1965 г., когда были доказаны положительные влияния на течение дисциркуляторной энцефалопатии. В дальнейшем были проведены исследования, подтвердившие эффективность этой группы препаратов в лечении когнитивных и других нарушений у пациентов с хронической ишемией мозга, инсультом, являющихся частой патологией, регистрируемой при СД [25, 38]. Доказано, что под влиянием сывороточных гемодериватовулучшаются мыслительные процессы, концентрация внимания, память, уменьшается интенсивность головной боли ишемически-гипоксического характера [25, 40]. Позитивный эффект CГД «Актовегин» подтвержден в многоцентровом рандомизированном исследовании при оценке состояния диабетической дистальной нейропатии, в котором доказано улучшение течения и уменьшение проявлений нейропатии при отсутствии негативного влияния препарата на углеводный обмен [44].
Актовегин содержит ряд основных микроэлементов – натрий, калий, кальций, фосфор, магний. Магний является каталитическим центром всех известных сегодня нейропептидов головного мозга и имеет статус нейроседативного иона, что имеет принципиальное значение для обеспечения нейропротективного эффекта.
Сывороточные гемодериваты эффективно используются в комплексной терапии мозгового инсульта (ишемического и геморрагического). При этом наряду со значимым уменьшением субъективных и объективных симптомов отмечается достоверное улучшение мозгового кровотока.
В результате лечения ишемического инсульта у 82% больных отмечено субъективное улучшение состояния, а у 61% – объективное уменьшение неврологической симптоматики [9]. При ишемических формах инсультов целесообразно назначать метаболическую терапию как можно раньше, так как в это время происходит активация перекисного окисления липидов при ишемии [3, 34]. Однако учитывая закономерности эксайтотоксичности, имеются рекомендации начинать введение нейропротекторов после 10–30-минутной реперфузии – по достижении максимального уровня продуктов перикисного окисления липидов в ишемическом очаге.
СГД используются при геморрагических инсультах, обеспечивая снижение медленно-волновой активности на ЭЭГ в области очага кровоизлияния, снижение риска возникновения сосудистого спазма, развития стойкого неврологического дефицита и последующей инвалидизации [28]. Достигнутые эффекты связывают с оксигенацией окружающих очаг тканей, снижением уровня лактата и нормализацией электрической активности нейронов коры. Поскольку сывороточные гемодериваты повышают потребление кислорода клетками, авторы не рекомендуют использовать его высокие дозы в острейшем периоде геморрагического инсульта, в сочетании с угнетением сознания. Лучший эффект использования СДГ при геморрагическом инсульте отмечается на 4–7-е сутки после внутримозгового кровоизлияния, когда уменьшаются явления отека мозга.
В восстановительном периоде инсульта сывороточные гемодериваты приводят к улучшению психических и когнитивных нарушений, обеспечивая улучшение речевых и двигательных функций, регресс психических симптомов беспокойства и возбуждения, нарушения поведения [20, 21].
Перечисленные эффекты позволяют прогнозировать эффективность сывороточные гемодериваты при дисциркуляторной энцефалопатии в отношении когнитивных (запоминание новой информации и воспроизведение уже имеющейся), двигательных и психических нарушений [23, 26, 31]. Отмечено субъективно значительное улучшение общего состояния у 44% пациентов, у 47% больных отмечен значимый регресс двигательных нарушений. Повышение интереса к пище, прогулкам, общению с окружающими отмечены у 66,7%; снижение тревожности, повышенной возбудимости – у 64,3%, улучшение качества сна – у 54,5%. Аффективные нарушения уменьшились у 67,9% пациентов, ипохондрия – у 60,6%.
Таким образом, сывороточные гемодериваты – универсальные препараты, которые могут быть использованы при сосудистых и метаболических поражениях головного мозга. Обладая антиоксидантными и другими метаболическими эффектами, СГД активизируют нейрональный метаболизм и микроциркуляцию, оказывают позитивное влияние на течение дистальной формы нейропатии и являются безопасными и не оказывают отрицательного влияния на метаболизм углеводов [22]. В то же время необходимо проведение исследований по оценке церебропротективного эффекта у пациентов с СД, осложненным различными циркуляторными нарушениями и при развитии когнитивных нарушений и деменции, обусловленных выраженными симптоматическими гипогликемиями.
Л И Т Е Р А Т У Р А
1. Левин О.С., Дамулин Н.В. Диффузные изменения белого вещества (лейкоapeoз) и проблема сосудистой деменции // Достижения в нейрогериатрии: сборник научных трудов / Под ред. Н.Н. Яхно и И.В. Дамулина. – М., 1995. – 293 с.
2. Шмырев В.И., Остроумова О.Д., Боброва Т.А. // Рус. мед. журн. – 2003. – Т. 11, № 4. – С. 216–220.
3. Янсен В., Брукнер Г.В. // Рус. мед. журн. – 2002. – Т. 10, № 12–13. – С. 543–546.
4. Abbott R.D., Donahue R.P., MacMahon S.W. et al. // JAMA. – 1987. – Vol. 257. – P. 949–952.
5. Air E.L., Kissela B.M. // Diabetes Care. – Vol. 30. – P. 3131–3140.
6. Akomolafe A., Beiser A., Meigs J.B. et al. // Arch. Neurol. – 2006. – Vol. 63. – P. 1551–1555.
7. Allen K.V., Frier B.M., Strachan M.W. // Eur. J. Pharmacol. – 2004. – Vol. 490. – P. 169–175.
8. Amiel S.A., Dixon T., Mann R., Jameson K. // Diabet. Med. – 2008. – Vol. 25, N 3. – P. 245–254.
9. Araki G. // Kiso to Rinsho. – 1974. – Vol. 8. – P. 4208–4214.
10. Biessels G.J., Staekenborg S., Brunner E. et al. // Lancet Neurol. – 2006. – Vol. 5. – P. 64–74.
11. Boyle P.J. // Diabetologia. – 1997. – Vol. 40, Suppl 2. – S69–S74.
12. Christopher T.K., Elizabeth R. // Endocr. Rev. – 2008. – Vol. 29, N 4. – P. 494–511.
13. Cryer P.E. // J. Clin. Invest. – 2007. – Vol. 117, N 4. – P. 868–870.
14. Cryer P.E. // Diabetologia. – 2002. – Vol. 45. – P. 937–948.
15. Cox D.J., Kovatchev B.P., Gonder-Frederick L.A. et al. // Diabet. Care. – 2005. – Vol. 28. – P. 71–77.
16. De Groot B.M., Machicao F. // Res. Comm. Chem. Path. Pharm. – 1990. – Vol. 68. – P. 125–128.
17. Derev’yannykh E.A., Bel’skaya G.N., Knoll E.A. et al. // Neurosci. Behav. Physiol. – 2008. – Vol. 38, N 8. – P. 873–875.
18. Festa A., D’Agostino R.J., Mykkanen L. et al. // Arterioscler. Thromb. Vasc. Biol. – 1999. – Vol. 19. – P. 562–568.
19. Gelling R.W., Morton G.J., Morrison C.D. et al. // Cell Metabolism. – 2006. – Vol. 3, N 1. – P. 67–73.
20. Herrmann W.M., Bohn-Olszewsky W.J., Kuntz G. // Z Geriatrie. – 1992. – Vol. 5. – P. 46–55.
21. Herrschaft H., Kunze U., Gleim F. // Med. Welt. – 1977. – Bd. 28. – S. 339–345.
22. Jacob S., Dietze G.J., Machicao F. et al. // Arzneimittelforschung. – 1996. – Bd. 46. – P. 269–272.
23. Jacobson A.M., Musen G., Ryan C.M. et al. // NEJM. – 2007. – Vol. 356. – P. 1842–1852.
24. Kane W.C., Aronson S.M. // Am. J. Pathol. – 1968. – Vol. 52. – P. 71–75.
25. Kanowski S., Kinzler E., Lehmann E. et al. // Pharmacopsychiatry. – 1995. – Vol. 28. – P. 125–133.
26. Klein K., Siedek H. // Med. Welt. – 1965. – Vol. 13. – P. 647–650.
27. Kume T., Asai N., Nishikava H. et al. // Proc. Natl. Acad. Sci. USA. – 2002. – Vol. 99. – P. 3288–3293.
28. Lencher H., Logar C., Grieshofer P. // New Trends in Clin. Neuropharmacol. – 1991. – Vol. 5. – P. 147–152.
29. Megherbi S.E., Milan C., Minier D. et al. // Stroke. – 2003. – Vol. 34. – P. 688–694.
30. Miyamoto E. // J. Pharmacol. Sci. – 2006. – Vol. 100. – P. 433–442.
31. Nagatsuka K., Tsuda Y., Takano T. et al. // Geriat. Med. – 2002. – Vol. 26. – P. 1202–1215.
32. Parsons C.G., Danysz W., Quack G. // Drug News Perspect. – 1998. – Vol. 11. – P. 523–569.
33. Peress N.S., Kane W.C. // Prog. Brain Res. – 1973. – Vol. 40. – P. 473–483.
34. Reeves M.J., Vaidya R.S., Fonarow G.C. et al. // Stroke. – 2010. – Vol. 41, N 5. – P. e409–e417.
35. Ryan C.M., Williams T.M., Finegold D.N., Orchard T . J. // Diabetologia. – 1993. – Vol. 36, N 4. – P. 329–334.
36. Sander D., Sander K., Poppert H. // Brit. Journ. Diabetes & Vasc. Disease. – 2008. – Vol. 8, N 5. – P. 222–229.
37. Schmidt R., Schmidt H., Fazekas F. // J. Neurol. – 2000. – Vol. 247. – P. 81–87.
38. Semlitsch H.V., Anderer P., Saletu B., Hochmayer I. // Neuropsychobiol. – 1990/91. – Vol. 24. – P. 49–56.
39. Stamler J., Vaccaro O., Neaton J.D. et al. // Diabetes Care. – 1993. – Vol. 16. – P. 434–444.
40. Talypov A.E., Ioffe I.S., Miatchin M.I., Kuksova N .S. // Zh. Nevrol. Psikhiatr. Im. S.S. Korsakova. – 2008. – Vol. 108, N 8. – P. 20–23.
41. Tuttolomondo A., Pinto D., Di Raimondo A., Licata G. // Diabetes Care. – 2007. – Vol. 30, N 11. – P. e114–e124.
42. Warren R.E., Frier B.M. // Diabetes Obes. Metab. – 2005. – Vol. 7, N 5. – P. 493–503.
43. Wright R.J., Frier B.M. // Diabetes Metab. Res. Rev. – 2008. – Vol. 24, N 5. – P. 353–363.
44. Ziegler D., Movsesyan L., Mankovsky B. et al. // Diabetes Care. – 2009. – Date of access: doi:10.2337/dc09-0545.
Медицинские новости. – 2011. – №6. – С. 15-18.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.