Внимание! Статья адресована врачам-специалистам
DotsenkoE.A.1, BobkovV.Ya.1, SolodovnikovaS.A.2, SemakI.V. 3, AlekseevN.A.3, ZharskayaS.M.3,
MalyushkovaE.V.3, RozhdestvenskiyD.А.4, IgnatchikI.О.1, Golyak А.А.1
1Belarusian State Medical University, Minsk, Belarus
2The 5-th Сity Сlinical Hospital, Minsk, Belarus
3Belarusian State Medical University, Minsk, Belarus
4Eurasian Economic Commission, Moscow, Russia
Bioequivalence of hypoglycemic medicinal products with modified release of GLIKLAZID MV and DIABETON MR in adult healthy volunteers
Резюме. Дана оценка биоэквивалентности, фармакокинетических профилей и безопасности лекарственного средства Гликлазид МВ, таблетки с модифицированным высвобождением 60 мг (СООО «Лекфарм», Республика Беларусь), в условиях их однократного и многократного приема здоровыми добровольцами после еды в сравнении с препаратом Диабетон MR 60 мг, таблетки с модифицированным высвобождением 60 мг (Les Laboratoires Servier, Франция). Представлены результаты открытого, рандомизированного, перекрестного исследования, которые показали сопоставимую биодоступность по полноте и скорости всасывания, что свидетельствует об полной биоэквивалентности препаратов.
Ключевые слова: биоэквивалентные испытания, гликлазид, модифицированное высвобождение.
Медицинские новости. – 2016. – №10. – С. 47–51.
Summary. The estimation of bioequivalence, pharmacokinetic profiles and safety of medicinal product Gliklazid MV with modified release of 60 mg (Lekpharm JLLC, Republic of Belarus) under the conditions of single and multi-dose application by healthy volunteers after meals in comparison with medicinal product Diabeton MR 60 mg, tablets with modified release of 60 mg (Les Laboratoires Servier, France) was given. The results of an open, randomized, crossover study, which showed comparable bioavailability of completeness and rate of absorption, which indicates their full bioequivalence of drugs, were presented.
Keywords: bioequivalence study, gliklazid, modified release.
Meditsinskie novosti. – 2016. – N10. – P. 47–51
Гликлазид – сульфонамидное производное мочевины, пероральный гипогликемический препарат, применяемый для лечения инсулинонезависимого сахарного диабета (II типа).
Основные механизмы действия гликлазида заключаются в увеличении секреции инсулина ?-клетками поджелудочной железы, а также повышении чувствительности ?-клеток к стимуляции глюкозой [2, 6, 18]. Гликлазид восстанавливает ранний пик инсулиносекреции и предотвращает избыточный выброс инсулина во второй фазе секреции, что наиболее близко к физиологическому профилю секреции инсулина. Также препарат повышает чувствительность тканей к инсулину. Гликлазид снижает риск тромбозов мелких сосудов, ингибируя агрегацию и адгезию тромбоцитов и снижая концентрацию факторов активации тромбоцитов; восстанавливает фибринолитическую активность сосудистого эндотелия и повышает активность тканевого активатора плазминогена; предупреждает развитие микроваскулитов, в том числе сетчатой оболочки глаза.
Гликлазид МВ – новая лекарственная форма гликлазида, содержащая специальный гидрофильный матрикс, регулирующий высвобождение действующего вещества, что позволяет принимать этот препарат перорально 1 раз в сутки. Прием пищи не влияет на абсорбцию гликлазида из пищеварительного тракта. Такая форма лекарственного средства (ЛС) дает лучший контроль над заболеванием и повышает комплаенс пациента.
Гликлазид МВ имеет линейные фармакокинетические параметры при дозировке 15–120 мг. После приема внутрь гликлазид полностью адсорбируется, концентрация его в плазме нарастает в течение 6 часов, уровень плато поддерживается от 6 до 12 часов. Гликлазид связывается с белками плазмы на 95%, метаболизируется в печени с образованием метаболитов, не обладающих гипогликемической активностью. Выводится преимущественно почками в виде метаболитов, менее 1% в неизмененном виде.
На фармацевтическом рынке нашей страны известен препарат Диабетон MR (Les Laboratoires Servier, Франция), что делает актуальной проблему разработки, оценки эффективности и безопасности отечественного препарата с замедленным высвобождением.
Цель исследования – оценка биоэквивалентности, фармакокинетических профилей и безопасности ЛС Гликлазид МВ, таблетки с модифицированным высвобождением 60 мг, производство СООО «Лекфарм», Республика Беларусь, в условиях их однократного и многократного приема здоровыми добровольцами после еды. Исследование выполнено по протоколу GCZD-LPHM-2012 (от 17.10.2012), разработанному в соответствии с требованиями [4, 8–10, 16, 17, 19].
Материалы и методы
ЛС.Тестируемое средство (Т) –Гликлазид МВ, таблетки с модифицированным высвобождением 60 мг (Лекфарм, Республика Беларусь), референтное (R) – Диабетон MR 60 мг, таблетки с модифицированным высвобождением 60 мг (Les Laboratoires Servier, Франция).
Этические аспекты. Клиническое испытание одобрено комитетом по этике 5-й городской клинической больницы г. Минска. Добровольцы были информированы о методах и условиях исследования, получено письменное согласие от всех добровольцев. Участие в испытаниях было добровольным.
Дизайн(рис. 1). Открытое рандомизированное перекрестное, в 4 периода и 4 последовательности, с перекрестным дизайном (RT/TR) в условиях однократного и многократного приема биоэквивалентное испытание у взрослых здоровых добровольцев в условиях приема после еды. Исследование включало 2 этапа: этап однократного и этап многократного приема ЛС. Отмывочный период между периодами составлял 2 недели. 30 здоровых добровольцев были рандомизированы на 2 равные группы (по 15 человек в каждой).
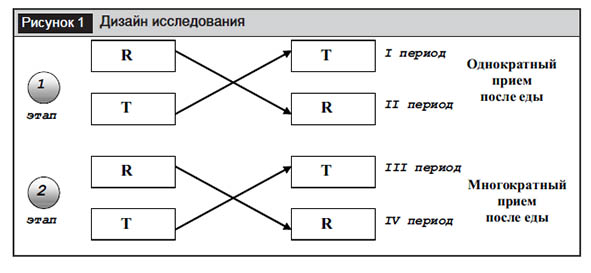
На 1-м этапе (однократный прием) добровольцы принимали ЛС в дозе 60 мг (1 таблетка тестируемого и референтного препаратов) однократно утром сразу после приема стандартного горячего завтрака с повышенным содержанием жира и калорий (общая калорийность 800–1000 ккал) с 200 мл воды. Добровольцы не употребляли жидкость в течение 2 часов после приема ЛС. В остальное время доброволец мог пить жидкость в объеме равном до 2 л в сутки. Во время пребывания в клиническом центре добровольцы обеспечивались 3-разовым питанием (в рамках стола Б), исключая кофеин-содержащие напитки и продукты, фруктовые соки.
На 2-м этапе (многократный прием ЛС) добровольцы в течение 1–6-го дней приема посещали клинический центр в амбулаторном режиме, где в тех же условиях принимали ЛС, имея возможность круглосуточной связи с врачом-исследователем при возникновении экстренной ситуации. На 7-й день приема добровольцы постоянно находились в клиническом центре. Продолжительность испытания для каждого добровольца составила 52 дня.
Критерии включения. В исследовании участвовали 30 здоровых добровольцев обоего пола, отобранных в соответствии со следующими критериями включения: возраст – 18–45 лет, индекс массы тела (ИМТ) 18,5–30 кг/м2, уровень гликемии натощак 4,9–6,0 ммоль/л. Диагноз «Здоров» у добровольцев устанавливали по результатам клинического, лабораторного и инструментального обследований.
Критерии невключения. Из исследования исключались лица с отягощенным аллергологическим анамнезом, острыми и хроническими заболеваниями, острыми инфекционными заболеваниями менее чем за 4 недели до начала испытания; с указаниями в анамнезе на применение любых ЛС менее чем за 2 недели до начала исследования, регулярное употребление алкоголя, курение, вегетарианство, участие в I фазе клинических испытаний ЛС менее чем за 3 месяца до начала испытания.
Обследование добровольцев и безопасность. На этапе скрининга проводился сбор демографических данных, анамнеза, физикальный осмотр с регистрацией основных показателей жизнедеятельности (температуры тела, частоты пульса, артериального давления – Ттела, ЧСС, АД), ЭКГ, клинический лабораторный скрининг: общий анализ крови (ОАК), биохимический анализ крови (БАК), общий анализ мочи (ОАМ); осуществляли серодиагностику сифилиса, ВИЧ-инфекции, гепатитов В и С, тест на беременность (для женщин).
С целью предотвращения развития гипогликемии добровольцы каждые 30 минут в течение первых 4 часов после приема ЛС выпивали 100 мл 10% раствора глюкозы. В период нахождения добровольца (1-й этап и 7-й день 2-го этапа) в исследовательском центре каждые 2 часа проводился клинический осмотр, измерение уровня глюкозы плазмы крови. При многократном приеме ЛС (2-й этап) контроль осуществлялся в амбулаторном режиме, при визите добровольцев в исследовательский центр.
Оценка безопасности применения ЛС (развитие ожидаемых и неожиданных побочных реакций) проводилась при каждом физикальном осмотре пациентов при пребывании их в клиническом центре, а также путем проведения ОАК, БАК, ОАМ через 24 часа при очередном отборе крови в амбулаторном режиме.
Сбор образцов крови. На 1-м этапе (однократный прием) отбор образцов крови в объеме 10 мл проводился до приема ЛС (0 ч), а также через 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 14, 24, 36, 48, 72 часа после приема ЛС.
На 2-м этапе (многократный прием) отбор образцов крови проводили до приема ЛС на 4–6-й дни приема; на 7-й день забор венозной крови осуществляли по аналогии с 1-м этапом.
Отбор образцов крови проводили путем пункции локтевой вены с установкой кубитального катетера. В течение 15 минут после получения крови пробирки центрифугировали в течение 10 минут при 3000 об./мин. Сыворотку крови после центрифугирования тотчас же консервировали в криокамере при температуре не выше минус 30°С.
Биоаналитическая процедура. Гликлазид из сыворотки крови выделяли путем твердофазной экстракции на неполярном сорбенте из слабокислой среды (рН около 3,5) с последующим элюированием 70% водным раствором ацетонитрила.
Количественное определение гликлазида в сыворотке крови проводили методом высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием с помощью методики, разработанной и полностью валидированной в соответствии с международными требованиями [14, 15, 21].
Хроматографическое разделение и масс-спектрометрический анализ проводили на жидкостномнг хроматографе LCMS-2020 (Shimadzu, Япония). Анализируемые соединения элюировали в градиентном режиме подвижной фазой: вода – ацетонитрил – 0,1 М ацетат аммония (рН 6,8) при температуре 40ºС.
Предел определения составил 5,0 нг/мл (процентная мера правильности 103 и 98%), предел обнаружения равен 0,5 нг/мл. Процентная мера правильности для всего диапазона применения методики при проведении измерений в один день составила 95–106%, в разные дни – 98–106%.
Фармакокинетические параметры [12, 22]. С учетом полученных уравнений регрессии и экспериментальных данных при однократном приеме ЛС рассчитывали базовые параметры: АUСt – площадь под фармакокинетической кривой до момента окончания исследования (72 часа) – рассчитывали по методу простых трапеций внемодельным способом; АUC??– площадь под фармакокинетической кривой (при условии экстраполирования кривой в бесконечность) – рассчитывали модельным способом с последующей логарифмической трансформацией данных.
Внемодельным способом по экспериментальным данным определяли также: Tmax – время достижения максимальной концентрации; Cmax – максимальная концентрация препарата в сыворотке крови. Методами нелинейного регрессионного анализа рассчитывали Сt и kel.
Для оценки констант фармакокинетического уравнения использовали метод наименьших квадратов с последовательным логарифмированием. Далее, используя пошаговый алгоритм точечной оценки отношения средних и 90% доверительных интервалов (ДИ), определяли параметры сравнительной биодоступности как отношение средних значений фармакокинетических параметров для тестируемого и референтного ЛС (f, f’, f”).
С учетом полученных уравнений регрессии и экспериментальных данных при многократном приеме ЛС рассчитывали базовые параметры: AUC?,ss – площадь под фармакокинетической кривой в течение 24-часового междозового интервала после установления стационарного распределения ЛС по методу простых трапеций. Внемодельным способом по экспериментальным данным определяли Cmax и Cmin – значения минимальной (в конце интервала дозирования) и максимальной (максимальная концентрация в пределах интервала дозирования) концентраций; f’ (AUC?,SS) – площадь под фармакокинетической кривой за время равное интервалу дозирования; f” (Cmax) – максимальная концентрации препарата в стационарную фазу.
Статистический анализ [1, 3, 23]. Ауксологические показатели добровольцев рассчитывали как среднее значение ± стандартное отклонение (Хср±SD). Все фармакокинетические параметры рассчитывались методами описательной статистики. Дисперсионный анализ (ANOVA), двойные односторонние тесты оценки биоэквивалентности, 90% ДИ для отношений среднего квадратического отклонения Т/R (тестируемое ЛС/референтное ЛС), оценка мощности и отношений проведены в отношении нетрансформированных и логарифмически трансформированных фармакокинетических параметров Cmax, AUC0-t.
Критерии эквивалентности. Гипотеза о биоэквивалентности испытуемого препарата и препарата сравнения принималась, если выполнялись условия:
1. 90% ДИ (при величине ошибки ?=0,05 или р=0,95 с двусторонней оценкой ДИ и мощности метода 1-?=0,8) для f, f’, f” находились в пределе 0,8–1,25.
2. Различия Cmax/AUCt и/или Cmax/AUC? недостоверны.
3. При однофакторном дисперсионном анализе показателей Сmax, AUCt и AUC? не выявлялись статистически достоверные различия как между препаратами, так и периодами их приема.
Результаты и обсуждение
Демографическая характеристика добровольцев. Демографическая характеристика добровольцев, участвовавших в биоэквивалентном исследовании, представлена в табл. 1. Среди 30 добровольцев женщин было 23, мужчин – 7. Средний возраст составил 33,3±1,42 года, рост – 171,1±1,67 см, вес – 71,3±2,31 кг; ИМТ – 24,2±0,55 кг/м2. В результате рандомизации добровольцы были разделены на 2 группы, демографические характеристики были сопоставимы. Все 30 добровольцев завершили 1-й этап, на 2-м этапе один доброволец отказался от участия без объяснения причин. Таким образом, 2-й этап (многократный прием) полностью завершили 29 добровольцев
Таблица 1. Демографическая характеристика добровольцев
|
Демографические показатели
|
Вся группа
|
Группа №1
|
Группа №2
|
|
Возраст, лет
|
33,3±1,42
|
32,9±2,00
|
33,6±2,24
|
|
Рост, см
|
171,1±1,67
|
168,1±1,45
|
174,4±3,12
|
|
Вес, кг
|
71,3±2,31
|
68,1±2,57
|
73,9±4,05
|
|
ИМТ, кг/м2
|
24,2±0,55
|
24,1±0,87
|
24,1±0,72
|
|
Мужчины/женщины
|
7/23
|
2/13
|
5/10
|
Фармакокинетика и биоэквивалентность при однократном приеме после еды. Средний профиль концентраций ЛС при однократном приеме после еды представлен на рис. 2, основные фармакокинетические параметры гликлазида – в табл. 2. В среднем Tmax составило для Гликлазида МВ 8,7±3,0 часа, для Дибетона MR – 9,1±2,7; Cmax – 1823,4±831,6 и 1933,5±779,3 нг/мл для Гликлазида МВ и Дибетона MR соответственно. Таким образом, статистически значимых различий в максимальных концентрациях гликлазида и временем ее достижения для 2 лекарственных препаратов не выявлено (p>0,05).
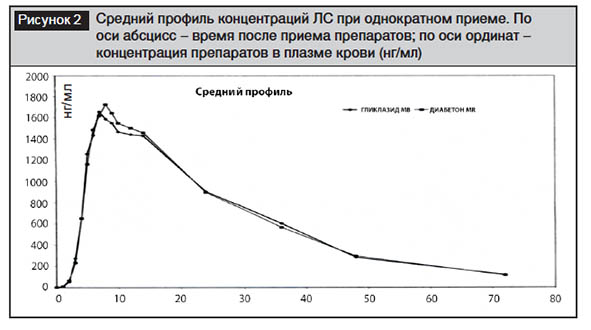
Таблица 2. Фармакокинетические параметры гликлазида при однократном приеме после еды
|
Параметр
|
Гликлазид МВ
|
Диабетон MR
|
|
Cmax, нг/мл
|
1823,4±831,6
|
1933,5±779,3 (p>0,05)
|
|
Tmax, ч
|
8,7±3,0
|
9,1±2,7 (p>0,05)
|
|
AUC72, нг?час/мл
|
45862,3±19914,9
|
46 066,3±14 611,9 (p>0,05)
|
|
AUC∞, нг?час/мл
|
47596,2±21232,0
|
47 651,8±15 657,9 (p>0,05)
|
|
T1/2,ч
|
8,6±1,8
|
8,8±1,8 (p>0,05)
|
Параметры биодоступности препаратов представлены в табл. 3. Следует обратить внимание, что для Cmax 90% ДИ соответствовал 82,7–102,5%, AUCt – 86,0–103,9%, для AUC∞ – 86,0–104,1%, что свидетельствует об отсутствии различий между референтным и тестируемым препаратами. Полученные нами данные близки к известным из литературы [20]: при приеме 30 мг гликлазида с модифицированным высвобождением 18 здоровыми добровольцами после еды Tmax составило 5,56–8,11 часа; Cmax была приблизительно в 2 раза меньше, чем при приеме гликлазида в дозе 60 мг (918,33–996,48 нг/мл); площадь под кривой была также приблизительно в 2 раза ниже: для AUC0–t 24 074–24 338 нг?час/мл, для AUC0–∞ 26 969–27 574 нг?час/мл. При приеме здоровыми добровольцами 60 мг таблеток гликлазида с модифицированным высвобождением [24] Cmax составила 2240–2410 мг/мл, Tmax – 8,40–9,1 часа, что близко к полученным нами данным.
Таблица 3. Показатели биоэквивалентности гликлазида (однократный прием)
|
Параметр
биодоступности (ln)
|
f”(CmaxT/CmaxR)
|
f’ (AUCtT/AUCtR)
|
f (AUC∞T/ AUC∞R)
|
|
90% ДИ
|
-0,190±0,025 82,7–102,5
|
-0,151±0,038 86,0–103,9
|
-0,151±0,041 86,0–104,1
|
Фармакокинетические параметры и биоэквивалентность при многократном приеме гликлазида после еды. Средний профиль концентраций двух препаратов представлен на рис. 3. Обращает на себя внимание сходный фармакокинетический профиль для обоих препаратов (табл. 4). Стационарное состояние на фоне многократного приема гликлазида с модифицированным высвобождением характеризовалось средней концентрацией (Css) 1476,9±630,0 нг/мл (для тестируемого) и 1549,8±886,7 нг\мл (для референтного). Полученная Css близка к известным из литературы: так, при приеме модифицированного гликлазида в дозе 60 мг в течение 6 дней его концентрация колебалась от 1300 до 1500 нг/мл [24].
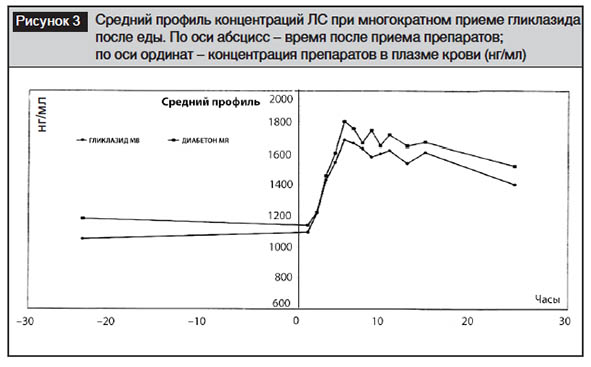
Таблица 4. Фармакокинетические параметры гликлазида в стационарном состоянии (при многократном приеме после еды)
|
Параметр
|
Гликлазид МВ
|
Диабетон MR
|
|
Css, нг/мл
|
1476,9±630,0
|
1549,8±886,7 (p>0,05)
|
|
Cmax, нг/мл
|
2272,5±773,3
|
2528,2±1257,5 (p>0,05)
|
|
Tmax, ч
|
7,0±3,8
|
6,9±3,6 (p>0,05)
|
|
AUC24, нг?час/мл
|
35445,1±15385,9
|
37 195,4±21 657,4 (p>0,05)
|
Cmax для обоих препаратов практически не различалась (2272,5±773,3 и 2528,2±1257,5 нг/мл для Гликлазида МВ и Диабетона MR соответственно). Полученные концентрации несколько меньше, чем известно из литературы [24]: 3000–3300 нг/мл. Tmax составило 7,0±3,8 и 6,9±3,6 часа для тех же препаратов соответственно.
Параметры биодоступности препаратов при многократном приеме ЛС представлены в табл. 5. Для Cmax 90% ДИ соответствовал 81,7–98,5%, AUCt – 90,7–106,1%, что свидетельствует об отсутствии различий между референтным и тестируемым препаратами.
Таблица 5. Показатели биоэквивалентности гликлазида в стационарном состоянии (многократный прием)
|
Параметр биодоступности
|
f” (CmaxT/CmaxR)
|
f’(AUCtT/AUCtR)
|
|
90% ДИ
|
-0,202±0,016
81,7–98,5
|
-0,097±0,059
90,7-106,1
|
Анализ мощности исследования, проведенный в соответствии с Государственной фармакопеей Республики Беларусь [4], показал, что мощность исследования по гликлазиду на выборке в 24 человека составила не менее 80% (табл. 6). Таким образом, объем выборки в 29 человек удовлетворяет значению рассчитанного диапазона.
Таблица 6. Анализ мощности исследования
|
Параметр
|
Однократный прием
|
Стационарное состояние
|
|
CV (%)
|
µT/µR
|
ß
|
n
|
CV (%)
|
µT/µR
|
ß
|
n
|
|
Cmax
|
22,0
|
0,94
|
=80
|
24
|
19,0
|
0,95
|
=80
|
19
|
|
AUCt
|
19,3
|
0,99
|
=80
|
16
|
15,8
|
0,95
|
=80
|
14
|
Безопасность добровольцев. ЛС Гликлазид МВ и Диабетон MR переносились хорошо, побочных реакций зарегистрировано не было.
Выводы:
1. ЛС Гликлазид MB, таблетки с модифицированным высвобождением 60 мг, производство СООО «Лекфарм», Республика Беларусь, и Диабетон MR 60 мг, таблетки с модифицированным высвобождением 60 мг, производство Les Laboratoires Servier, Франция, являются биоэквивалентными, поскольку для действующего вещества – гликлазида – выполняются критерии сопоставимости оценки сравнительной биодоступности – ДИ для величин f, f’, f” при однократном приеме, f’, f” при многократном приеме не превышают диапазон 80,0–125,0% по максимальной концентрации в сыворотке крови, площадям под фармакокинетическими кривыми в условиях однократного приема прандиально и многократного приема.
2. Мощность исследования по активному фармакологическому ингредиенту – гликлазиду – достигла не менее 80% для выборки объемом 1–24 добровольца.
3. ЛС Гликлазид MB и Диабетон MR 60 мг обладают эквивалентными параметрами скорости абсорбции, поскольку для действующего вещества отсутствуют статистически достоверные различия между отношениями максимальной концентрации гликлазида к площади в условиях однократного приема прандиально и многократного приема.
4. Нежелательные эффекты при приеме ЛС Гликлазид MB и Диабетон MR отсутствовали.
Л И Т Е Р А Т У Р А
1. Бондарева И.Б. // Клинич. фармакокинетика. – 2004. – №1. – С.14–22.
2. Викис А.Д. // Украiнський мед. часопис. –2002. – №3 (29). – С.6–15.
3. Гланц С. Медико-биологическая статистика / С. Гланц. – М., 1999. – 460 с.
4. Государственная фармакопея Республики Беларусь (ГФ. РБ II). Т.1. Общие методы контроля качества лекарственных средств. 5.8. Биодоступность и биоэквивалентность генерических лекарственных средств / М-во здравоохр. Респ. Беларусь, УП «Центр экспертиз и испытаний в здравоохранении»; под общ. ред. А.А. Шерякова – Молодечно, 2012. – С.1092–1130.
5. Каркищенко Н.Н., Хоронько В.В., Сергеева С.А., Каркищенко В.Н. Фармакокинетика. – Ростов н/Д., 2001. – 384 с.
6. Корпачев В.В. // Украiнський мед. часопис. – 2002. – №3 (29). – С.16–22.
7. Проведение качественных исследований биоэквивалентности лекарственных средств. Метод. указания / И.Б. Бондарева, В.Б. Герасимов, А.П. Дрожжин и др.: утв. Министерством здравоохранения и социального развития Российской Федерации 10.08.2004 г.
8. Проведение качественных исследований биоэквивалентности лекарственных средств. Метод. указания / Под ред. В.Г. Кукеса, А.А. Фирсова, А.К. Стародубцева, В.П. Жердева: Утверждены МЗ РФ 10.08.04. – М., 2004. – 43 с.
9. Рождественский Д.А. // Рецепт. – 2008. – №6 (62). – С.28–34.
10. Руководство 42-7.1:2005 «Руководство по клиническим исследованиям. Лекарственные средства. Исследование биодоступности и биоэквивалентности». – Киев, 2005.
11. Сергиенко В.И. Математическая статистика в клинических исследованиях / В.И. Сергиенко, И.Б. Бондарева. – М., 2000. – 256 с.
12. Соколов А.В. // Клинич. фармакокинетика. – 2004. – №1. – С.5–13.
13. CPMP/EWP/QWP/1401/98 Rev.1/Corr Guideline on the Investigation of the Bioequivalence. – London, 2010. – P.1–27.
14. EMEA/CHMP/EWP/192217/2009 Guideline on Validation of Bioanalytical Methods. – London, 2009.
15. Guidance for Industry, Bioanalytical Method Validation, US Department of Health and Human Services, Food and Drug Administration Centre for Drug Evaluation and Research (CDER). Centre for veterinary Medicine (CVM). – 2001.
16. Investigation of Bioavailability and Bioequivalence. – Commission of the European Communities. – 1991. – III/54/89-EN.– P.1–20.
17. In vivo Bioequivalence Guidances. – U.S. Pharmacopeia 24-NF 19< Supplement 2. – 2000. – 1090, P.2056–2098.
18. Michael A. // Circ. Res. – 2008. – №102. – Р.164–176.
19. Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability / WHO Technical Report Series. – 2006. – N937. – P.347–390.
20. Rojanasthien N., Autsavakitipong T., Kumsorn B., Manorot M., Teekachunhatean S. // International Scholarly Research Network ISRN Pharmacology. – 2012. – Vol.2012. – 6 p.
21. Sharma V.K., Mazumder B. // World J. Pharmaceutical Res. – 2014. – Vol.3, Iss.9. – P.979–997.
22. Wagner J.G. // J. Pharmacokinet. Biopharm. – 1976. – Vol.4, N3. – P.281–285.
23. Yamaoka K., Nakagawa T., Uno T. // J. Pharmacokinet. Biopharm. – 1978. – Vol.6, N6. – P.547–558.
24. Yang J.-F., Wei G.-L., Lu R., et al. // Asian J. Pharmacodynamics Pharmacokinetics. – 2006. – Vol.6, N2. – P.153–160.
Медицинские новости. – 2016. – №10. – С. 47-51.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.