Внимание! Статья адресована врачам-специалистам
Omonova U.T.
Tashkent Pediatric Medical Institute, Uzbekistan
Distribution of dystrophin gene deletions in Uzbekistan population
Резюме. Представлены данные молекулярно-генетических исследований 91 пациента с мышечной дистрофией Дюшенна/Беккера из 81 семьи. Результаты исследований показали, что в популяции Узбекистана преобладают семейные случаи прогрессирующей мышечной дистрофии Дюшенна/Беккера над мутацией de novо, а также протяженные делеции в 3 ,-конце гена дистрофина. По результатам косвенной ДНК-диагностики, в семьях высокого риска в популяции Узбекистана наиболее информативными являются внутригенные высокополиморфные маркеры STR45 и STR49.
Ключевые слова: прогрессирующая мышечная дистрофия, молекулярно-генетические исследования, дистрофин.
Медицинские новости. – 2017. – №10. – С. 42–44.
Summary. The article presents data of molecular genetic studies among 91 patients with Duchenne/Becker muscular dystrophy from 81 families. The results of the research showed that in the population of Uzbekistan the family cases of progressive Duchenne/Becker muscular dystrophy over the de novo mutation prevail, as well as the long deletions in the 3’dion of the dystrophin gene. Based on the results of indirect DNA diagnostics in high-risk families in the population of Uzbekistan, the most highly informative are the highly polymorphic markers STR45 and STR49.
Keywords: progressive muscular dystrophy, molecular-genetic studies, dystrophin.
Meditsinskie novosti. – 2017. – N10. – P. 42–44.
Мышечная дистрофия Дюшенна (G71.0 Мышечная атрофия; синонимы – псевдогипертрофическая миодистрофия Дюшенна, прогрессирующая мышечная дистрофия Дюшенна, болезнь Дюшенна, паралич Дюшенна) – самая злокачественная форма миодистрофий и наиболее распространенная. Популяционная частота данного заболевания: 1:3500 мальчиков и 21,7 на 100 000 живорожденных мальчиков [3, 5]. В группу дистрофинопатий включены прогрессирующие мышечные дистрофии Дюшенна и Беккера (МДД/Б), которые представляют собой аллельные варианты, обусловленные различными мутациями в гене белка дистрофина [1, 3]. Приблизительно 55–65% всех случаев МДД/Б обусловлены делециями гена дистрофина различной протяженности, 5–10% случаев – дупликациями части гена, у остальных имеют место точковые мутации [2, 4].
В настоящее время накоплены обширные данные в отношении делеционных спектров в гене дистрофина в различных популяциях мира, установлены определенные популяционные различия [2, 5]. До настоящего времени молекулярно-генетическое исследование МДД/Б в Республике Узбекистан не проводилось, поэтому для разработки подходов пренатальной, дифференциальной, пресимптоматической диагностики и определения носительства мутантного гена актуальным является изучение молекулярно-генетической природы МДД/Б в Узбекистане.
Цель исследования – изучить генетический полиморфизм гена дистрофина среди больных с мышечной дистрофией Дюшенна/Беккера и определить частоту носительства мутации гена дистрофина у родственниц в семьях больных с МДД/Б в популяции Узбекистана.
Материалы и методы
Молекулярно-генетические исследования выполнялись в Научно-исследовательском институте гематологии и переливания крови Министерства здравоохранения Республики Узбекистан в отделе молекулярной медицины и клеточных технологий. Молекулярно-генетические исследования проводились по двум направлениям: мультиплексная ПЦР-диагностика для выявления мажорных делеций у больных с МДД/Б и проведение косвенной ДНК-диагностики для определения носительства мутантного гена дистрофина у родственниц больного с МДД/Б. Геномную ДНК из образцов периферической крови (Vacutainer Becton Dickinson International с ЭДТА) выделяли с использованием набора для выделения ДНК DIAtom™ DNA Prep100 ООО «Центр молекулярной генетики» (Российская Федерация) в соответствии с инструкцией. Амплификацию проводили с помощью термоциклеров GeneAmp PCR-system 2720 (Applied Biosystems, США) и Corbett Palm Cycler (Corbett Research, модель СG1-96, Австралия) с использованием наборов DMD-del и DMD-CA (ООО «Центр молекулярной генетики», Российская Федерация).
Прямая ДНК-диагностика проведена 91 пациенту с МДД/Б из 81 семьи, у 84 (92,3%) из них – мышечная дистрофия Дюшенна, у 7 (8,6%) – мышечная дистрофия Беккера. Анализ проводился по 20 экзонам гена дистрофина – промоторная область, 3, 4, 6, 8, 13, 17, 19, 32, 42, 43, 44, 45, 47, 48, 50, 51, 52, 53, 60 экзоны (рис. 1).

Косвенная диагностика проведена в 21 семье, отягощенной МДД/Б, с применением внутригенных высокополиморфных маркеров, расположенных в 45-м (STR45), 49-м (STR49) и 50-м (STR50) интронах гена.
Результаты и обсуждение
Исследуемая патология имеет Х-сцепленный рецессивный тип передачи, основная группа семей состоит в неродственных браках и прямой корреляции заболевания с частотой родственных браков не было выявлено. По результатам генеалогического опроса обнаружено 5 семей близкородственных браков, что составило около 6,2% от общего количества обследуемых, делеции выявлены в 4 (80%) случаях близкородственных браков. Соответственно неродственные браки составили 93,8% (76 семей), делеции выявлены в 59,2% (45 семей) случаев (рис. 2).
Все мутации гена дистрофина, обнаруженные в 4 семьях с родственными браками, характеризовались делециями одного экзона в дистальном районе 3’-конца (делеции 48, 50 и 53 экзонов), причем в 3 семьях мышечная дистрофия Дюшенна впервые определялась в поколении.
По результатам молекулярной диагностики у 33 (36,3%) пациентов из 32 (39,5%) семей делеции не обнаружены, а у 58 (63,7%) пациентов из 49 (60,5%) семей были выявлены мажорные делеции гена дистрофина различной длины – от одного до девяти экзонов: в 65,3% семьях верифицировались протяженные делеции, делеции одного экзона встречались в 34,7% семей. Основной делеционный спектр располагался в дистальной части гена дистрофина – 3’-конце (делеции 40–60 экзонов), что составило 81,6% (40 семей, 47 пациентов) (рис. 3).
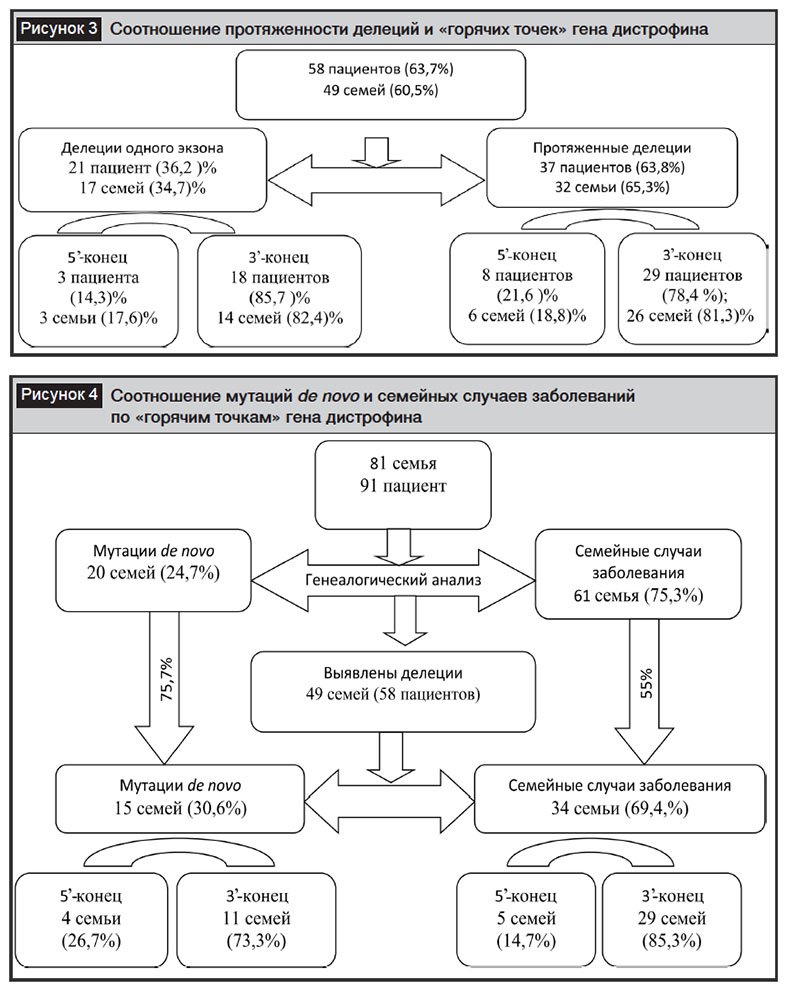
При анализе делеций наиболее часто выявлялись в дистальной части гена дистрофина, что составляло 81,6% от всех выявленных мутаций: 50 экзон – в 24 случаях, 51 экзон – в 24 случаях, 52 экзон – в 21 случае, 48 экзон – в 16 случаях, 53 экзон – в 14 случаях; делеций промоторной зоны, 32, 42 и 60 экзона не было выявлено (рис. 4).
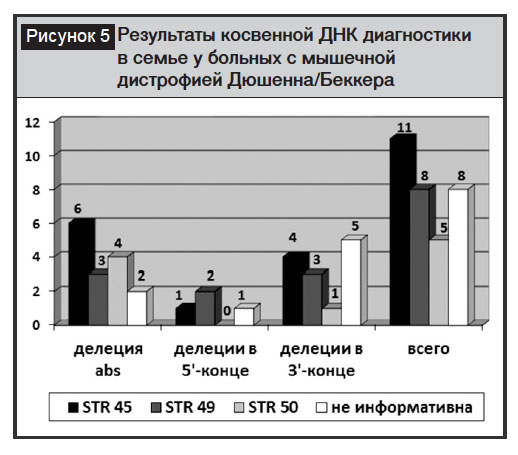
Косвенная диагностика проведена в 21 семье, отягощенной прогрессирующей мышечной дистрофией Дюшенна/Беккера, с применением внутригенных высокополиморфных маркеров, расположенных в STR45, STR49, STR50 интронах гена. В 33,3% (7 семей) случаев при выполнении прямой ДНК-диагностики «мажорные» делеции в гене дистрофина не были выявлены, в 14,3% (3 семьи) – обнаружены делеции в 5’-конце, в 52,4% (11 семей) – выявлены делеции в 3’-конце.
Провели 81 косвенную ДНК-диагностику в 21 семье высокого риска по МДД/Б: из обследованных семей 8 (38,1%) не были информативны по STR45, STR49, STR50; в 13 (61,9%) семьях у родственниц пробанда с МДД/Б был подтвержден статус носительства мутации гена дистрофина (рис. 5).
На базе Республиканского центра «Скрининг матери и ребенка» внедрен метод пренатальной инвазивной диагностики ПМД Дюшенна/Беккера. Было обследовано 6 семей высокого риска по данной патологии, в 5 случаях проведен кордоцентез, в 1 – амниоцентез.
Таблица 1. Спектр делеций гена дистрофина
|
Номер
экзона
|
Делеции
1 экзона
(21 пациент,
17 семей)
|
Протяженные
делеции
(37 пациентов,
32 семьи)
|
ВСЕГО
|
|
3
|
–
|
2
|
2
|
|
4
|
–
|
3
|
3
|
|
6
|
1
|
4
|
5
|
|
8
|
–
|
4
|
4
|
|
13
|
–
|
4
|
4
|
|
17
|
–
|
6
|
6
|
|
19
|
2
|
6
|
8
|
|
32
|
Делеции
не выявлены
|
Делеции
не выявлены
|
Делеции
не выявлены
|
|
42
|
Делеции
не выявлены
|
Делеции
не выявлены
|
Делеции
не выявлены
|
|
43
|
7
|
1
|
8
|
|
44
|
–
|
1
|
1
|
|
45
|
–
|
3
|
3
|
|
47
|
1
|
9
|
10
|
|
48
|
1
|
15
|
16
|
|
50
|
3
|
21
|
24
|
|
51
|
1
|
23
|
24
|
|
52
|
3
|
18
|
21
|
|
53
|
2
|
12
|
14
|
|
60
|
Делеции
не выявлены
|
Делеции
не выявлены
|
Делеции
не выявлены
|
|
Промоторная зона
|
Делеции
не выявлены
|
Делеции
не выявлены
|
Делеции
не выявлены
|
По результатам ДНК-диагностики выявлено 3 плода мужского пола с идентифицированными делециями в гене дистрофина и рекомендовано прерывание беременности по медико-генетическим показаниям. В остальных семьях беременность была пролонгирована, проведено постнатальное обследование рожденных детей с целью подтверждения отсутствия поражения в гене дистрофина.
Выводы:
1. Результаты собственных молекулярно-генетических исследований показали, что в популяции Узбекистана преобладают семейные случаи ПМД Дюшенна/Беккера над мутацией de novо, а также протяженные делеции в 3’-конце гена дистрофина, что соответствует международным литературным данным.
2. По данным мультиплексной ПЦР пациентов с мышечной дистрофией Дюшенна выявлены мутации в 16 экзонах из 20 исследованных, что дает основание для использования набора для прямой ПЦР-диагностики «мажорных» делеций в гене дистрофина в популяции Узбекистана.
3. По результатам косвенной ДНК-диагностики в семьях высокого риска в популяции Узбекистана наиболее информативными являются внутригенные высокополиморфные маркеры STR45 и STR49.
Л И Т Е Р А Т У Р А
1. Грознова О.С., Шаховская М.С., Артемьева С.Б., Тренева Н.И. // Российский вестник перинатологии и педиатрии. – 2011. – Т.56, №3. – С.46–48.
2. Дадали E.Л., Мальмберг С.А., Подагова Е.В., Поляков A.B., Петрухин A.C. // Российский медицинский журнал. – 2007. – №3. – С.18–21.
3. Евтушенко С.К., Шаймурзин М.Р., Евтушенко Л.Ф. // Таврический медико-биологический вестник. – 2009. – Т.12, №2. – С.46.
4. Шаймурзин М.Р., Евтушенко С.К. // Вестник физиотерапии и курортологии. – 2010. – №6. – С.40–41.
5. Bertini E. // Semin. Pediatr. Neurol. – 2011. – Vol.18, N4. – P.277–288.
Медицинские новости. – 2017. – №10. – С. 42-44.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.