Внимание! Статья адресована врачам-специалистам
Trisvetova E.L.
Belarusian State Medical University, Minsk
Vascular protection in arterial hypertension –
step to reduce the risk of cardiovascular complications
Резюме. Одним из звеньев патогенеза артериальной гипертензии является ремоделирование артериальных сосудов, усугубляющее течение заболевания, способствующее раннему поражению и прогрессированию органных нарушений, развитию ассоциированных клинических состояний. Применение эффективных комбинированных препаратов (Престанс – периндоприл и амлодипин), влияющих на состояние артериальных сосудов, способствует улучшению эластических свойств магистральных артерий, уменьшает структурное ремоделирование сосудов, снижает сердечно-сосудистую заболеваемость и смертность.
Ключевые слова: артериальная гипертензия, ремоделирование сосудов, жесткость сосудистой стенки, антигипертензивные препараты, периндоприл, амлодипин, Престанс.
Медицинские новости. – 2017. – №11. – С. 3–7.
Summary. One of the links of the pathogenesis of arterial hypertension is remodeling of arterial vessels, aggravating the course of the disease, contributing to early injury and progression of organ disorders, development of associated clinical conditions. The use of effective combination drugs (Prestans – perindopril + amlodipine), affecting the condition of arterial vessels, improves the elastic properties of the main arteries, reduces structural remodeling of vessels, reduces cardiovascular morbidity and mortality.
Keywords: arterial hypertension, vascular remodeling, vascular wall stiffness, antihypertensive drugs, perindopril, amlodipine, Prestans.
Meditsinskie novosti. – 2017. – N11. – P. 3–7.
По результатам многих клинических международных исследований доказано влияние уровня артериального давления (АД) и сопутствующих факторов риска на сердечно-сосудистые, почечные осложнения и смертность пациентов с артериальной гипертензией (АГ) [1]. Оценка сердечно-сосудистого риска при АГ наряду с факторами риска, клинически манифестными сердечно-сосудистыми заболеваниями, сахарным диабетом и хронической болезнью почек включает бессимптомное поражение органов-мишеней, возникающее в результате изменения сосудистого русла [2]. К одному из звеньев патогенеза АГ относят функциональную и морфологическую перестройку артериальных сосудов – ремоделирование, усугубляющее течение заболевания. Изменения сосудистого русла, кровоснабжающего головной мозг, сердце, почки, способствуют раннему поражению и прогрессированию органных нарушений при АГ, обусловливают развитие ассоциированных клинических состояний [3]. Современное представление о течении, прогнозе АГ, выборе и эффективности влияния антигипертензивных лекарственных средств включает оценку состояния артериальных сосудов.
При длительном повышении АД сосудистые изменения на первом этапе проявляются функциональными признаками, включающими сосудосуживающие реакции, на втором – морфологической перестройкой, характеризующейся утолщением медиального слоя стенки и уменьшением просвета сосуда. Сужение или спазм сосудов обусловливает нарушение транспортной, проводящей, функции артериальной системы. Структурная перестройка артерий крупного и среднего калибра у пациентов с АГ представлена гипертрофией и/или гиперплазией гладкомышечных клеток, увеличением толщины медиального слоя стенки сосуда, нередко повреждением эндотелия и уменьшением просвета сосуда [4].
В стенках аорты и магистральных сосудов изменяется структура внеклеточного матрикса, содержащего эластиновые и коллагеновые волокна. Волокна эластина определяют механические характеристики сосудов в условиях низкого давления, а фибриллы коллагена, жесткость которых в 10–100 раз превышает таковую у эластиновых, – при высоком давлении. При АГ происходит уменьшение соотношения эластин/коллаген, увеличение объема внеклеточного матрикса соединительной ткани [5]. Морфологические изменения стенок вызывают снижение эластических свойств сосудов, ухудшающих тем самым демпфирующую функцию артериальной системы. Демпфирующие свойства артериальной системы необходимы для превращения пульсирующего артериального кровотока, продуцируе-мого ритмическими сокращениями сердца, в непрерывный ламинарный на уровне капилляров [6].
При сохраненной эластичности сосудистой стенки артериальная пульсовая волна, формирующаяся из двух составляющих – прямой (движется от сердца к периферии) и отраженной (движется от периферии к сердцу), поддерживает адекватный уровень АД [6]. Ухудшение эластических свойств артерий сопровож-дается повышением систолического АД вследствие выброса крови в ригидную артериальную систему. Повышение систолического АД вызывает увеличение постнагрузки левого желудочка и развитие гипертрофии миокарда, повышение потребности миокарда в кислороде, появление диастолической дисфункции и хронической сердечной недостаточности [7].
В случае ригидности артериальной стенки создаются условия, изменяющие скорость распространения пульсовой волны (СРПВ) и продолжительность прямой и отраженной пульсовой волны. В фазу систолы волна давления отражается в любом участке артериального русла, вызывая отраженную волну, распространяющуюся в обратном направлении к восходящей аорте. При нормальной эластичности артериальных стенок и низкой СРПВ отраженная волна возвращается в восходящую аорту в период диастолы, повышая диастолическое АД и не изменяя уровень систолического АД [6]. В случае жесткости артериальной стенки и возрастания СРПВ отраженная волна возвращается раньше, во время систолы, что сопровождается повышением пикового конечного систолического давления в восходящей аорте и снижением диастолического АД, определяющего коронарную перфузию. Снижение перфузии миокарда – фактор, инициирующий коронарную недостаточность. Ухудшение кровоснабжения сердца при увеличении жесткости артериальных сосудов является одним измеханизмов развития ишемии миокарда и сердечной недостаточности [7, 8].
Повреждение артериальных сосудов прогрессирует на фоне повышения систолического и пульсового АД, замыкая «порочный круг», нарушается функция системы микроциркуляции с развитием патологической морфологической перестройки тканей и органов [9].
Таким образом, снижение эластичности сосудистой стенки и повышение жесткости аорты и крупных артерий, перестройка микроциркуляторного русла с развитием аневризм, кровоизлияний обусловливают повреждение органов-мишеней при АГ, тем самым увеличивая риск развития сердечно-сосудистых осложнений и смерти (рис. 1). Согласно результатам исследований, снижение эластичности и повышение жесткости артериальной стенки является независимым прогностическим фактором, повышающим риск развития фатальных и нефатальных сердечно-сосудистых событий, смертности от общих причин у пациентов с АГ [8, 10].
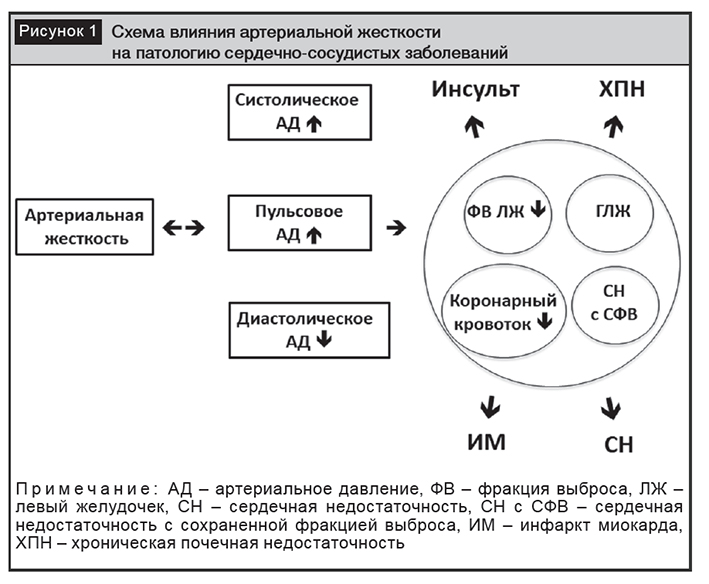
Снижение эластичности и жесткость сосудистой стенки увеличиваются с возрастом и зависят от многих факторов, в числе которых генетические, регулирующие синтез и катаболизм коллагена и эластина, степень выраженности и прогрессирования атеросклероза и артериолосклероза [5].
В клинической практике для оценки жесткости сосудистой стенки применяют неинвазивный воспроизводимый метод измерения СРПВ. Обычно волны регистрируют чрескожно на правой общей сонной артерии и правой бед-ренной артерии. СРПВ рассчитывают, исходя из времени запаздывания (?t) [6, 7].
В большинстве эпидемиологических исследований, анализировавших прогностическую ценность СРПВ, изучали три основных артериальных участка: аортальный ствол (carotid-femoral), артерии верхних (carotid-brachial) и нижних конечностей (femoral-dorsalis pedis). Помимо СРПВ применяют оценку других показателей, характеризующих жесткость артериальных сосудов: центральное аортальное давление, центральное пульсовое давление, индекс ригидности артерий, плече-лодыжечный индекс, сердечно-лодыжечный индекс (CAVI), индекс аугментации (Alx), время распространения отраженной волны, индекс эффективности субэндокардиального кровотока [1, 7]. Вместе с тем, скорость каротидно-феморальной пульсовой волны считают основным доступным для практического здравоохранения методом измерения артериальной жесткости.
Поскольку артериальная жесткость нередко предшествует клинической манифестации сердечно-сосудистых событий и является предиктором смертности пациентов с АГ, при определении тактики лечения следует учитывать влияние антигипертензивного препарата на СРПВ [10, 11].
K.T. Ongи соавт. представили доказательства влияния антигипертензивных препаратов на артериальную жесткость независимо от уровня снижения АД, выполнив мета-анализ15 рандомизированных контролируемых двойных слепых параллельных групповых исследований [12]. Первичной конечной точкой считали изменения скорости феморально-бедренной пульсовой волны на фоне лечения у 294 пациентов с умеренной АГ. Рандомизированные группы пациентов получали ингибиторы АПФ (n=75), блокаторы кальциевых каналов (n=75), ?-адреноблокаторы (n=30), диуретики (n=26) и плацебо (n=88). В краткосрочном (до28 дней) и долгосрочном (более 28 дней) наблюдении в группах с антигипертензивным лечением СРПВ снизилась на 0,75 и -1,3 м/с по сравнению с группой плацебо (+0,17 и -0,44 м/с). При краткосрочном лечении отмечено более выраженное уменьшение артериальной жесткости в группе получавших ингибиторы АПФ, при долгосрочном – улучшение наблюдали во всех группах с антигипертензивным лечением по сравнению с показателями у лиц, получавших плацебо.
Исследователи отметили, что снижение артериальной жесткости не зависело от уровня снижения АД.
Клинические исследования, выполненные за истекшее десятилетие, доказали прогностическую значимость исследования СРПВ в оценке сердечно-сосудистого риска. A. Scuteriи соавт., изучив результаты 17 проспективных исследований с участием 15 877 пациентов с оценкой прогностической ценности СРПВ в аорте, показали, что увеличение показателя скорости на каждый 1 м/с ассоциировано с повышением общей смертности на 15%, сердечно-сосудистой смертности – на 14%, риска развития любого сердечно-сосудистого осложнения – на 15% [13].
Дальнейшее изучение влияния антигипертензивного лечения на прогноз и риск возникновения осложнений, ремоделирование сосудов у пациентов с АГ позволило установить вариативность показателей артериальной жесткости при применении различных антигипертензивных препаратов.
Результаты исследованияASCOT (Anglo-ScandinavianCardiacOutcomesTrial) с участием пациентов с АГ и высоким сердечно-сосудистым риском доказали преимущество использования комбинации ингибитора АПФ периндоприла с блокатором кальциевых каналов (БКК) амлодипином по сравнению с ?-адреноблокатором атенололом и тиазидным диуретиком бендрофлуметиазидом. Значительное снижение частоты сердечно-сосудистых событий: на 13% нефатального инфаркта миокарда и всех случаев смерти от ИБС; на 24% смерти от сердечно-сосудистых событий; на 23% фатальных и нефатальных инсультов; на 13% случаев нефатальных инфарктов миокарда; на 16% всех сердечно-сосудистых событий и процедур реваскуляризации наблюдали в группе, получавшей периндоприл и амлодипин [14].
Для изучения возможных механизмов действия и причин большей эффективностикомбинации периндоприла и амлодипина по сравнению с атенололом ибендрофлуметиазидом проведен анализ результатов фрагмента исследования ASCOT-CAFE (Conduit Artery Function Evaluation). У 2199 пациентов с АГ изучили показатели артериальной жесткости, центрального АД до и после лечения периндоприлом и амлодипином, атенололом ибендро-флуметиазидом.
Уровень снижения АД на плечевой артерии на фоне лечения не отличался, однако отметили, что в группе ингибитора АПФ и БКК снизились значительнее центральное систолическое АД (на 4,3 мм рт. ст.; р<0,0001), центральное аортальное пульсовое давление (на 3,0 мм рт. ст.; р<0,0001) по сравнению с показателями в группе, получавшей ?-адреноблокатор и диуретик. Не выявили различий в показателях СРПВ, вместе с тем более высокий уровень центрального систолического АД в группе пациентов, принимавших ?-адреноблокатор и диуретик, свидетельствовал о различном влиянии комбинаций препаратов на свойства отраженной волны (рис. 2). Замедление частоты сердечных сокращений и/или периферическая вазоконстрикция при приеме ?-адреноблокатора могла негативно влиять на отраженную волну. Более высокое значение (на 6,5%) Alx, отражающего процент прироста систолического АД при лечении ?-адреноблокатором и диуретиком, также свидетельствовало об измененныххарактеристиках отраженной волны [15].
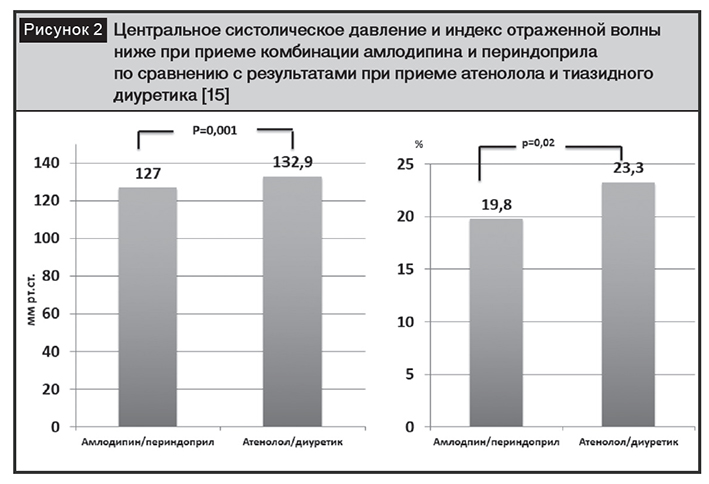
Таким образом, выявлены различия между классами антигипертензивных препаратов по влиянию на амплификацию АД. Ингибиторы АПФ и БКК (дигидропиридиновые) оказывают более благоприятное влияние на АД по сравнению с ?-адреноблокаторами и тиазидными диуретиками [16]. Ингибиторы АПФ и БКК, обладающие вазодилатирующим действием, расширяют артерии среднего и мелкого калибра, уменьшают гипертрофию мышечного слоя стенки, тем самым влияют на интенсивность волны, отраженной от периферического отдела сосудистого русла, а также на аугментацию и величину центрального АД [17, 18].
Также отмечено благоприятное влияние на жесткость артериальной стенки и регресс гипертрофии миокарда левого желудочка комбинации периндоприла и индапамида по сравнению с результатами лечения атенололом.
В клиническом исследованииREASON (REgression of Arterial Stiffness in a contrOlled double-bliNd) у пациентов с АГ изучили влияние периндоприла в комбинации с индапамидом на гипертрофию левого желудочка и жесткость артериальной стенке путем оценки СРПВ[19]. В течение одного года одна группа получала низкодозовую комбинацию периндоприла и индапамида (2 мг и 0,625 мг соответственно), другая – атенолол в дозе 50–100 мг. Комбинированная терапия периндоприлом и индапамидом в низких дозах привела к достоверному снижению скорости отраженной волны в сонной артерии, снижению центрального систолического и пульсового АД. Значительное уменьшение массы миокарда левого желудочка отметили в группе периндоприла и индапамида (-16,3 г) по сравнению с группой получавших атенолол (-4,3 г), при этом показатель массы миокарда не зависел от уровня снижения систолического, диастолического и среднего АД на плечевой артерии, а зависел от уровня центрального АД [20].
Рациональная комбинация ингибитора АПФ и дигидропиридинового БКК широко применяется в клинической практике, на основании результатов многих клинических исследований, в которых показан синергизм и эффективность компонентов в лечении АГ и ассоциированных состояний. В силу отличий ингибиторов АПФ по химической структуре и фармакокинетике препараты класса различаются по влиянию на клинические проявления сердечно-сосудистых заболеваний, профилактику осложнений, снижение риска развития смерти. В трех крупных исследованиях с использованием оригинального препарата периндоприла (EUROPA, PREAMI, ASCOT) доказаны очевидные различия препаратов класса ингибитора АПФ по первичной конечной точке и смертности. При этом вклад периндоприла в снижение риска составил 18% против 5% для других ингибиторов АПФ [14, 22, 23].
Периндоприл – липофильный ингибитор АПФ длительного действия, с высокой системной биодоступностью, способный проникать в ткани. Большое значение имеет тропность периндоприла к тканевой АПФ, поскольку ингибирование АПФ в органах (сердце, сосуды, почки) и тканях по сравнению со снижением уровня фермента в плазме крови при длительном лечении определяет фармакологические эффекты препарата. За счет высокого сродства к АПФ эндотелия и адвентиции сосудов, прочности связи с АПФ периндоприл не только снижает АД, но и улучшает функцию эндотелия и потокозависимую дилатацию артерий, уменьшает прогрессирование атеросклероза [24].
Под влиянием периндоприла, с одной стороны, замедляется образование или высвобождение вазоконстрикторных и антинатрийуретических веществ (альдостерон, норадреналин, аргинин-вазопрессин, эндотелин-1), с другой стороны, увеличивается содержание в тканях и крови вазодилатирующих и натрийуретических веществ (брадикинин, простагландины Е2 и I2, эндотелиальный фактор расслабления, предсердный натрийуретический пептид).
Периндоприл является единственным ингибитором АПФ с документированным эффективным влиянием на функцию сосудов в различных сегментах артериального дерева: эндотелиальную функцию коронарных и периферических артерий у пациентов с АГ, ремоделирование малых артерий при АГ, эластичность сонных артерий у пациентов с АГ и сахарным диабетом 2-го типа, скорость распространения пульсовой волны при АГ и у выживших пациентов с терминальной стадией хронической болезни почек (рис. 3) [25].
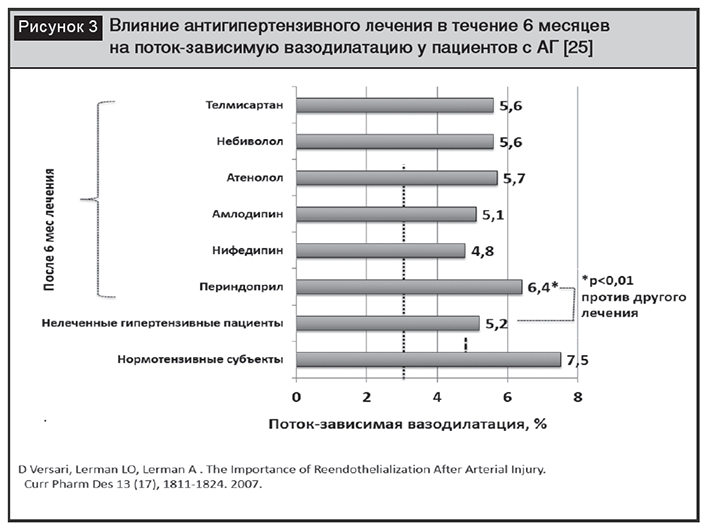
Анализ рандомизированных двойных слепых клинических исследований с участием периндоприла демонстрирует эффективность препарата в снижении и контроле АД у пациентов с АГ, обратном развитии нарушений сосудистой стенки и, в итоге, предотвращении сердечно-сосудистых событий. Долгосрочное лечение периндоприлом обусловливает уменьшение гипертрофии сосудистой стенки, улучшение функционального состояния мелких артерий, увеличение коронарного кровотока и коронарного резерва одновременно с регрессом периартериального и интерстициального коллагена стенок коронарных артериол. На фоне лечения периндоприлом уменьшается гипертрофия стенок сонной и лучевой артерии, уменьшается внутренний диаметр сонной артерии, что способствует улучшению эластичности сосудов, отчетливо увеличивается коронарный кровоток. У пациентов с терминальной стадией почечной недостаточности периндоприл уменьшает СРПВ независимо от изменений АД, что приводит к снижению риска смерти от всех причин и сердечно-сосудистых заболеваний [25].
Доказательная база эффективной защиты от развития сердечно-сосудистых осложнений и смерти комбинацией периндоприла и амлодипина, полученная во многих клинических рандомизированных исследованиях, послужила обоснованием для создания препарата Престанс (LesLaboratoriesServier, Франция). Фиксированная комбинация периндоприла аргинина и амлодипина безилата обусловлена их комплементарным механизмом действия, а также доказательной базой эффективной защиты от развития сердечно-сосудистых осложнений и смерти, полученной во многих клинических рандомизированных исследованиях.
Амлодипин относится к наиболее изученным антигипертензивным препаратам класса дигидропиридиновых БКК и оказывает выраженное дилатирующее действие на периферические и коронарные сосуды. Препятствуя трансмембранному току ионов кальция в гладкомышечные клетки сосудов и кардиомиоцитов и обладая большей селективностью к гладкомышечным клеткам сосудов, амлодипин снижает периферическое сосудистое сопротивление, не влияя на проводящую систему и не ухудшая сократительную способность миокарда [26, 27].
В многочисленных клинических исследованиях показано, что амлодипин, помимо снижения АД, проявляет антиатерогенное и антиоксидантное, регулирующее действие на синтез и распад оксида азота и функцию эндотелия, воздействует на пролиферацию гладкомышечных клеток и внеклеточного матрикса [28–30]. Гемодинамические эффекты амлодипина обусловливают улучшение кровоснабжения и микроциркуляции жизненно важных органов – головного мозга и почек.
В эксперименте и клинике доказан синергизм влияния амлодипина и ингибиторов АПФ. Установлено, что в результате блокирования деградации кининов ингибиторами АПФ и стимуляцией амлодипином образования кининов через калликреиновый путь, комбинация амлодипина и ингибитора АПФ приводит к наибольшему NO-зависимому снижению дефицита кислорода в миокарде [31, 32].
Препарат Престанс (доступные дозы 5/5, 5/10, 10/5 и 10/10 мг) предназначен для лечения пациентов с АГ, в том числе высокого риска, с целью улучшения прогноза и уменьшения симптомов заболевания. Оба компонента в препарате Престанс проявляют синергизм действия в отношении антигипертензивного эффекта: вазодилатирующие свойства БКК и релаксация сосудов вследствие блокады медленных кальциевых каналов потенцируются вазодилатацией, обусловленной ингибированием ангиотензина IIв плазме и тканях, вызванного подавлением АПФ периндоприлом. Усиление антигипертензивного действия сопровождается уменьшением частоты возникновения нежелательных реакций каждого компонента: отеки голеней, вызванные амлодипином, кашель, возникающий при приеме периндоприла. В результате использования фиксированной комбинации периндоприла и амлодипина – Престанса усиливаются сосудистые эффекты: уменьшается пролиферация и воспалительные изменения в сосудистой стенке, повышается антиоксидантная защита и замедляется прогрессирование атеросклероза, тромбоза, замедляется ремоделирование сосудов, улучшается структура сосудистой стенки и эндотелиальная функция, улучшается коронарный и почечный кровоток, повышается фибринолитический баланс крови [33].
Исследования патогенеза морфологических и функциональных изменений сердечно-сосудистой системы при АГ показали, что помимо снижения и контроля АД, антигипертензивные препараты должны проявлять дополнительные сосудистые эффекты. Разные классы антигипертензивных препаратов отличаются по влиянию на сосудистую стенку, артериальную жесткость, СРПВ, являющуюся одним из независимых факторов поражения органов-мишеней и риска развития сердечно-сосудистых осложнений и ассоциированных состояний. В клинических исследованиях представлены доказательства положительного влияния на артериальное сосудистое русло, уменьшение жесткости сосудистой стенки ингибиторов АПФ и БКК, независимое от уровня снижения АД. Применение комбинации препаратов на ранних стадиях заболевания способствует улучшению эластических свойств магистральных артерий, уменьшению структурного ремоделирования сосудов и центрального АД, следовательно, снижению сердечно-сосудистой заболеваемости и смертности.
ЛИТЕРАТУРА
1. 2013 ESC/ESC Guidelines for the management of arterial hypertension the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). – J. Hypertens. – 2013. – Vol.31, N7. – P.1281–1357.
2. Mancia G., De Backer G., Dominiczak A., et al. // J. Hypertens. – 2007. – Vol.25, N6. – P.1105–1187.
3. Lewington S., Clarke R., Qizilbash N., et al. // Lancet. – 2002. – Vol.360. – P.1903–1913.
4. Struijker-Boudier H.A., Cohuet G.M., Baumann M., et al. // J. Hypertens. Suppl. – 2003. – Vol.21, N3. – S19–S23.
5. Laurent S., Boutouyrie P., Lacolley P. // Hypertension. – 2005. – Vol.45, N6. – P.1050–1055.
6. Eur. Heart J. – 2010. – Vol.31, N19. – P.2338–2350.
7. Laurent S., Cockcroft J., Van Bortel L., et al. // Eur. Heart J. – 2006. – Vol.27, N21. – P.2588–2605.
8. Kingwell B.A., Waddell T.K, Medley T.L., et al. // JACC. – 2002. – Vol.40, N4. – P.773–779.
9. Levy B.I., Ambrosio G., Pries A.R., et al. // Circulation. – 2001. – Vol.104, N6. – P.735–740.
10. Boutouyrie P., Tropeano A.l., Asmar R., et al. // Hypertension. – 2002. – Vol.39, N1. – P.10–15.
11. Volpe M., Battistoni A., Tocci G., et al. // J. Hypertens. – 2012. – Vol.30, N6. – P.1056–1064.
12. Ong K.T., Delerme S., Pannier B., et al. // J. Hypertens. – 2011. – Vol.29, N6. – P.1034–1042.
13. Scuteri A., Tesauro M., Appolloni S., et al. // J. Hypertens. – 2007. – Vol.25, N5. – P.1035–1040.
14. Dahlof B., Sever P.S., Poulter N.R., et al. // Lancet. – 2005. – Vol.366. – P.895–906.
15. Williams B., Lacy P.S., Thom S.M., et al. // Circulation. – 2006. – Vol.113, N9. – P.1213–1225.
16. Protogerou A.D., Stergiou G.S., Vlachopoulos C., et al. // Curr. Pharm. Des. – 2009. – Vol.15, N3. – P.272–289.
17. Morgan T., Lauri J., Bertram D., et al. // Am. J. Hypertens. – 2004. – Vol.17, N2. – P.118–123.
18. Pannier B.M., Guerin A.P., Marchais S.J., et al. // Clin. Exp. Pharmacol Physiol. – 2001. – Vol.28, N12. – P.1074–1077.
19. Asmar R.G., London G.M., O’Rourke M.E., et al. // J. Hypertens. – 2001. – Vol.38, N4. – P.922–926.
20. De Luca N., Asmar R.G., London G.M., et al. // J. Hypertens. – 2004. – Vol.22, N80. – P.1623–1630.
21. Niu W., Qi Y. // Int. J. Cardiol. – 2016. – Vol.1, N218. – P.109–117.
22. Fox K.M. // Lancet. – 2003. – Vol.362. – P.782–788.
23. Ferrari R. // Arch. Intern. Med. – 2006. – Vol.166, N6. – P.659–666.
24. Ceconi C., Fox K.M., Remme J.M., et al. // Cardiovasc. Res. – 2007. – Vol.73, N1. – P.237–246.
25. Laurent S. // J. Hypertens. – 2005. – Vol.18. – 155S–162S.
26. Versari D., Lerman N.O., Lerman A., et al. // Curr. Pharm. Des. – 2007. – Vol.13, N17. – P.1811–1824.
27. Toyo-oka T., Nayler W. // Blood Pressure. – 1996. – Vol.5, N4. – P.206–208.
28. Sarsero D., Fujiwara T., Molenaar P.,et al. // Br. J. Pharmacol. – 1998. – Vol.125, N1. – P.109–119.
29. ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. // JAMA. – 2002. – Vol.288, N23. – P.2981–2997.
30. Brown M.J., Palmer C.R., Castaigne A., et al. // Lancet. – 2000. – Vol.356. – P.366–372.
31. Batova S., DeWever J., Godfraind T., et al. // Cardiovasc. Res. – 2006. – Vol.71, N3. – P.478–485.
32. Mital S., Magneson A., Loke K.E., et al. // J. Cardiovasc. Pharmacol. – 2000. – Vol.36. – P.248–254.
33. Bahl V.K., Jadhav U.M., Thacker Н.Р. // Am. J. Cardiovasc. Drugs. – 2009. – Vol.9, N3. – P.135–142.
Медицинские новости. – 2017. – №11. – С. 3-7.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.