Внимание! Статья адресована врачам-специалистам
KostiukS.A.1, SimirskiV.V.1, GorbichY.L.2, AniskoL.A.3, PoluyanO.S.1
1Pilot Production of the Institute of Bioorganic Chemistry National Academy of Sciences of Belarus, Minsk
2Belarusian Medical Academy of Post-Graduate Education, Minsk
3City Clinical Infectious Diseases Hospital, Minsk, Belarus
Cytokine storm at COVID-19
Резюме. В статье рассматриваются иммунный ответ клеток, пораженных SARS-CoV или MERS-CoV, механизм возникновения, а также стратегии лечения индуцируемого вирусом SARS-CoV-2 чрезмерного и пролонгированного цитокинового/хемокинового ответа, известного как цитокиновый шторм при COVID-19. Во время пандемии коронавирусная инфекции (COVID-19) ухудшение состояния некоторых пациентов тесно связано с цитокиновым штормом, который рассматривается как одна из основных причин острого респираторного дистресс-синдрома и полиорганной недостаточности, что приводит к физиологическому истощению и смерти. Своевременный контроль цитокинового шторма на ранних его стадиях посредством иммуномодуляторов и антагонистов цитокинов, а также уменьшение инфильтрации воспалительных клеток в легкие играет ключевую роль в улучшении показателей эффективности лечения и снижении уровня смертности у пациентов с COVID-19.
Ключевые слова: SARS-CoV-2, COVID-19, цитокиновый шторм.
Медицинские новости. – 2020. – №10. – С. 4–8.
Summary. The article discusses the immune response of cells affected by SARS-CoV or MERS-CoV, the mechanism of occurrence, and treatment strategies for the SARS-CoV-2 virus-induced excessive and prolonged cytokine / chemokine response, known as the cytokine storm in COVID-19. During a coronavirus infection pandemic (COVID-19), the deterioration of some patients is closely associated with the cytokine storm, which is considered one of the main causes of acute respiratory distress syndrome and multiple organ failure, which leads to physiological exhaustion and death. Timely control of the cytokine storm in its early stages through immunomodulators and cytokine antagonists, as well as a decrease in the infiltration of inflammatory cells into the lungs, plays a key role in improving treatment efficacy and reducing mortality in patients with COVID-19.
Keywords: SARS-CoV-2, COVID-19, cytokine storm.
Meditsinskie novosti. – 2020. – N10. – P. 4–8.
Основу компонента неспецифической реакции организма на повреждающие воздействия – системного гуморального ответа – составляет активация экспрессии и, как следствие, повышение содержания в крови различных биологически активных веществ, прежде всего цитокинов. Цитокины представляют собой низкомолекулярные (8–50 кДа) пептиды (в основном гликопротеиды), которые могут продуцироваться различными типами клеток, преимущественно моноцитами, тканевыми макрофагами (система фагоцитирующих мононуклеаров), лимфоцитами, клетками ретикулоэндотелиальной системы, эндотелием, полиморфно-ядерными лейкоцитами, главным образом нейтрофилами [1, 3, 4].
Цитокины в исключительно малых нанопикомолярных концентрациях осуществляют гуморальную регуляцию межклеточных и межсистемных взаимодействий, определяют функциональную активность отдельных клеток, их способность к пролиферации и дифференцировке, выживаемость или программированную апоптотическую гибель. Повышение, особенно значительное, уровня цитокинов в плазме крови знаменует развитие системного гуморального цитокинового ответа организма на воздействие вируса [1, 4, 15].
Ответ клеток-мишеней на цитокины, которые, по сути, являются сигнальными молекулами, начинается с активации специализированных (цитокиновых) мембраносвязанных рецепторов, многие из которых уже идентифицированы, клонированы и начали использоваться в практической медицине в аспекте цитокиновой терапии [2].
Каждый цитокин, как правило, вызывает несколько различных эффектов однонаправленного (например, провоспалительного, иммуностимулирующего, апоптотического и т.д.) действия. При развитии цитокинового ответа образуются как цитокины-синергисты, усиливающие и пролонгирующие одни и те же эффекты, так и цитокины-антагонисты, ограничивающие или блокирующие эти же самые эффекты [1, 4].
Равновесное состояние преформированных пулов цитокинов с противоположным типом действия является одним из условий сохранения гомеостаза интактного организма, а адекватные друг другу однонаправленные изменения – увеличение цитокинов-антагонистов при развитии цитокинового ответа – обусловливают особенность реагирования организма на воздействия вирусов [1, 4, 15, 32].
Патогенетически значимый дисбаланс компонентов цитокинового ответа – это путь к неблагоприятному сценарию (по интенсивности, качеству и/или продолжительности) развития вирусной инфекции (рис. 1).
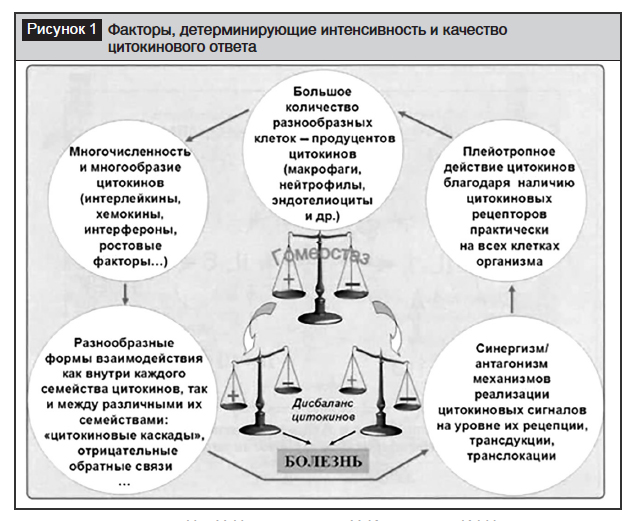
Способность цитокинов индуцировать образование друг друга, то есть формировать «цитокиновые каскады», предопределяет не только возможность амплификации и пролонгирования цитокиновых эффектов, но может явиться основой для включения патогенетически значимых для организма «порочных кругов». Организм включает защитно-приспособительные процессы – лихорадку, защитный иммунный ответ, воспаление, активацию гемопоэза, повышающие его резистентность к вирусу. При этом адекватность реагирования в значительной степени определяется согласованным взаимодействием стресс-реакции и цитокинового ответа [1, 14, 15, 51].
Данные, полученные при наблюдении за пациентами, перенесшими коронавирусную инфекцию, свидетельствуют о том, что в механизме развития инфекций, вызываемых коронавирусом, важную роль играет противовоспалительный ответ [34, 50].
Эксперименты на клеточных культурах in vitro показали, что в эпителиальных клетках дыхательных путей, дендритных клетках и макрофагах происходит отсроченное высвобождение цитокинов и хемокинов на ранней стадии вирусной инфекции, обусловленной SARS-CoV. Позже клетки выделяют противовирусные интерфероны в низких концентрациях и провоспалительные цитокины (интерлейкин (ИЛ)-1?, ИЛ-6, фактор некроза опухолей (ФНО)) и хемокины (хемокиновый (с ССмотивом) лиганд 2 (CCL2), CCL3 и CCL5) в высоких концентрациях [13, 29, 30].
Подобно вирусу, вызывающему тяжелый респираторный синдром (SARS-CoV), вирус ближневосточного респираторного синдрома (MERS-CoV) поражает эпителиальные клетки дыхательных путей человека, THP-1 клетки (клеточная линия моноцитарной лейкемии), макрофаги моноцитарного происхождения периферической крови, дендритные клетки и индуцирует отсроченное повышение уровня противовоспалительных цитокинов и хемокинов [52, 57]. После заражения MERS-CoV для выделения больших количеств противовирусных интерферонов индуцируются плазмоцитоидные дендритные клетки, а не мононуклеарные макрофаги [45]. Повышенные уровни содержания цитокинов и хемокинов в сыворотке крови у пациентов с MERS связаны с большим количеством нейтрофилов и моноцитов в легочных тканях и периферической крови, что позволяет предположить, что эти клетки могут играть ведущую роль в патологии легких [27, 33, 35].
Сходные явления наблюдались у пациентов с инфекцией SARS-CoV. Выработка противовирусных интерферонов ИФН 1-го типа (ИФН-???) является основополагающим звеном врожденного иммунного ответа на вирусные инфекции, а ИФН-1 – ключевая молекула, играющая антивирусную роль на ранних стадиях заболевания [1, 10]. Отсроченное высвобождение ИФН на ранних стадиях инфекций, обусловленных SARS-CoV и MERS-CoV, не способствует антивирусному ответу организма [10]. Впоследствии быстро повышающийся уровень цитокинов и хемокинов привлекает многие воспалительные клетки, такие как нейтрофилы и моноциты, что в результате приводит к избыточной инфильтрации клеток воспаления в ткани легких, вызывая их повреждение [9, 15].
Научные исследования свидетельствуют о том, что неуправляемый и/или чрезмерный цитокиновый/хемокиновый ответ клеток, пораженных SARS-CoV или MERS-CoV, может играть важную роль в патогенезе этих заболеваний [28, 38, 39, 55].
Экспериментальные модели на животных указывают на роль цитокинов и хемокинов в воссоздании легочной иммунопатологии после инфицирования коронавирусом человека HCoV. Несмотря на сходные титры вируса в дыхательных путях, у старых приматов, инфицированных SARS-CoV, с большей вероятностью разовьется нарушение регуляции иммунного ответа, чем у молодых животных, приводя к более серьезному проявлению заболевания [47]. Вероятно, чрезмерный воспалительный ответ в большей степени, чем титр вируса, может послужить причиной гибели старых приматов. Также у инфицированных SARS-CoV мышей линии BALB/c степень тяжести заболевания старых особей зависит от ранней и непропорционально сильной активации генов воспаления, имеющей отношение к острому респираторному дистресс-синдрому [43]. Быстрая репликация вирусов SARS-CoV у мышей линии BALB/c приводит к отсроченному высвобождению ИФН-???, что сопровождается миграцией многих «воспалительных» мононуклеарных макрофагов [11]. Накопившиеся мононуклеарные макрофаги получают сигналы активации посредством рецепторов ИФН-??? на их поверхности и вырабатывают больше моноцитарных хемоатрактантов (CCL2, CCL7 и CCL12), что, в свою очередь, приводит к дальнейшей аккумуляции моноцитарных макрофагов, которые повышают концентрацию провоспалительных цитокинов (ФНО, ИЛ-6, ИЛ-1?), утяжеляя таким образом течение заболевания [34].
Истощение воспалительных моноцитов-монофагов или нейтрализация цитокина воспаления ФНО защищало мышей от смертельной инфекции SARS-CoV. Кроме того, ИФН-??? или провоспалительные цитокины, вырабатываемые мононуклеарными макрофагами, индуцируют апоптоз Т-клеток, что в дальнейшем препятствует клиренсу вируса [11].
Еще одним следствием быстрой репликации вируса и интенсивного провоспалительного ответа цитокинов/хемокинов является индукция апоптоза клеток эпителия и эндотелия легких. ИФН-??? и ИФН-? активизируют инфильтрацию клеток воспаления посредством механизмов, вовлекающих Fas-лиганд (FasL) или ФНО-зависимый лиганд, индуцирующий апоптоз (TRAIL-death receptor 2 (DR5)), и способствуют апоптозу эпителиальных клеток дыхательных путей и альвеол легких [23, 24, 44]. Апоптоз клеток эндотелия и эпителия является причиной повреждения барьеров, создаваемых эпителиальными клетками капилляров легких и альвеол, вызывает транссудацию и альвеолярный отек легких и в итоге ведет к гипоксии. Следовательно, медиаторы воспаления играют ключевую роль в патогенезе острого дистресс-синдрома, который является основной причина смерти пациентов от коронавирусной инфекции, обусловленной SARS-CoV или MERS-CoV [18, 26].
Стало известно, что несколько провоспалительных цитокинов (ИЛ-6, ИЛ-8, ИЛ-1?, гранулоцитарномакрофагальный колониестимулирующий фактор, активные формы кислорода) и хемокинов (CCL2, CCL5, ИФН-?-индуцированный протеин 10 (IP10), CCL3) вносят свой вклад в возникновение острого респираторного дистресс-синдрома. Результаты научных исследований поддерживают точку зрения, что при коронавирусной инфекции SARS-CoV высокие титры вируса и несбалансированный цитокиновый/хемокиновый ответ являются причиной возникновения шторма воспалительных цитокинов [8, 25, 42].
Известно, что новая коронавирусная инфекция COVID-19 вызывает у заболевших цитокиновый шторм [34, 50].
Цитокиновый шторм, или гиперцитокинемия, – это потенциально летальная реакция иммунной системы, характеризуемая быстрой пролиферацией и повышенной активностью T-клеток, макрофагов и естественных киллеров с высвобождением защитными клетками различных воспалительных цитокинов и химических медиаторов [1, 6, 14, 15, 51]. Суть состояния состоит в выработке большого количества медиаторов воспаления, которые приводят к активации иммунных клеток и высвобождению последними новой порции медиаторов вследствие наличия неконтролируемой положительной обратной связи между этими процессами. Порочный круг вызывает разрушение тканей очага воспаления, одновременно реакция распространяется на соседние ткани и по мере развития приобретает системный характер, охватывая весь организм в целом. Цитокиновый шторм, как системную воспалительную реакцию в организме, бывает сложно различить на фоне исходного заболевания из-за схожести протекающих процессов. Из клинических особенностей могут присутствовать лихорадка с постоянной температурой, спленомегалия, гепатомегалия с нарушением работы печени, лимфаденопатия, коагулопатия, цитопения, кожная сыпь и различные неврологические симптомы [9, 46, 50].
Цитокиновый шторм сопровождается иммунопатологическими изменениями в легких. У пациентов с COVID-19 определяются высокие уровни экспрессии ИЛ-1?, ИФН-?, IP-10 и моноцитарного хемоаттрактантного белка 1 (MCP-1). Эти воспалительные цитокины могут активировать клеточный ответ, вызываемый Т-хелперами 1-го типа [31]. Активация Т-хелперов 1 – ключевое событие для разворачивания специфического иммунитета [12]. Однако в отличие от пациентов с SARS лица с COVID-19 имеют также повышенные уровни секретируемых Т-хелперами 2 цитокинов, таких как ИЛ-4 и ИЛ-10, которые ингибируют воспалительный ответ. Содержание в сыворотке крови ИЛ-2R и ИЛ-6 у пациентов с COVID-19 положительно коррелирует со степенью тяжести заболевания [56].
Другие исследования показали, что в сравнении с лицами с COVID-19 из отделения общей терапии пациенты из отделения интенсивной терапии имеют повышенные уровни гранулоцитарномакрофагального колониестимулирующего фактора, IP-10, MCP-1, воспалительного белка макрофагов 1А и ФНО-?. Изложенные выше результаты исследований позволяют утверждать, что цитокиновый шторм имеет положительную корреляцию с тяжестью заболевания [31].
Тяжело протекающая пневмония, вызванная новым типом коронавируса, у пациентов пожилого возраста с острым респираторным дистресс-синдромом характеризуется более высокими показателями смертности [41]. Основными патологическими изменениями при остром респираторном дистресс-синдроме является повреждение легочных и интерстициальных тканей по причине неспецифической инфильтрации воспалительных клеток [17].
Избыточное местное высвобождение цитокинов – решающий фактор, который индуцирует патологические изменения и клиническое проявление заболевания [37]. При этом сывороточные уровни цитокинов значительно повышены у пациентов с данной патологией, и степень их повышения положительно коррелирует с уровнем смертности [54].
Цитокиновый шторм также является ключевым фактором, определяющим клиническое течение внелегочной полиорганной недостаточности. Это частично объясняет признаки внелегочной функциональной недостаточности органов (повышение содержания печеночных ферментов и креатинина), наблюдаемые у некоторых пациентов с COVID-19 без дыхательной недостаточности, что позволяет предположить, что цитокиновый шторм является причиной повреждений тканей и органов, не относящихся к легким [48].
Таким образом, цитокиновый шторм является причиной острого респираторного дистресс-синдрома и/или внелегочной полиорганной недостаточности и важным фактором обострения течения COVID-19 и даже смерти.
Японские ученые выяснили, что именно может способствовать развитию цитокинового шторма, который приводит к острому респираторному дистресс-синдрому у пациентов с COVID-19 [34, 46, 50]. На данный момент известно, что SARS-CoV-2 проникает в клетки человека при помощи рецептора ACE2 и фермента TMPRSS2. Однако цитокиновый шторм и острый респираторный дистресс-синдром развиваются в организме на более поздних стадиях, даже в том случае, когда количество вируса может уменьшаться.
Учитывая эти взаимосвязи, японские ученые из университета Хоккайдо предположили, что воспаление должно начинаться по какому-то другому пути [46]. Когда SARS-CoV-2 проникает в здоровые клетки, то это уменьшает в них количество ACE2, что приводит к увеличению в крови полипептида ангиотензина II. Оказалось, что ангиотензин II запускает воспалительный путь NF -?B и ИЛ-6-STAT3 в не иммунных, а эндотелиальных и эпителиальных клетках. В результате это приводит к развитию цитокинового шторма, переходящего в острый респираторный дистресс-синдром.
«Усиление этого воспалительного пути может возникать с возрастом, что объясняет повышенный риск смертности от COVID-19 у пожилых людей», – заявил соавтор исследования Масааки Мураками [46].
Когда тяжелый острый респираторный синдром коронавируса поражает клетки, экспрессирующие поверхностные рецепторы ангиотензинпревращающего фермента 2 (АПФ-2) и TMPRSS2, активная репликация и высвобождение вируса заставляют клетку-хозяина высвобождать связанные с повреждением молекулярные структуры, включая АТФ, нуклеиновые кислоты и олигомеры ASC. Они распознаются соседними эпителиальными и эндотелиальными клетками и альвеолярными макрофагами, вызывая генерацию провоспалительных цитокинов и хемокинов (включая ИЛ-6, IP-10, макрофагальный воспалительный белок 1? (MIP-1?), MIP-1? и MCP-1) [34, 50]. Эти белки привлекают моноциты, макрофаги и Т-клетки к месту инфекции, способствуя дальнейшему воспалению.
ИЛ-6 может передавать сигналы по двум основным путям, которые называются классической цис-передачей или транс-передачей. При передаче цис-сигналов ИЛ-6 связывается с мембраносвязанным рецептором ИЛ-6 (mIL-6R) в комплексе с gp130; нисходящая сигнальная трансдукция опосредуется, в свою очередь, JAKs (Janus kinases) и STAT3 (сигнальный преобразователь и активатор транскрипции-3). Связанный с мембраной комплекс gp130 экспрессируется повсеместно в отличие от mIL-6R, экспрессия которого ограничена в основном иммунными клетками. Активация передачи сигналов в рамках цис-передачи приводит к плейотропным эффектам на приобретенный иммунитет (В- и Т-клетки), а также врожденную иммунную систему – нейтрофилы, макрофаги и естественные клетки-киллеры, которые могут вносить вклад в возникновение цитокинового шторма (рис. 2).
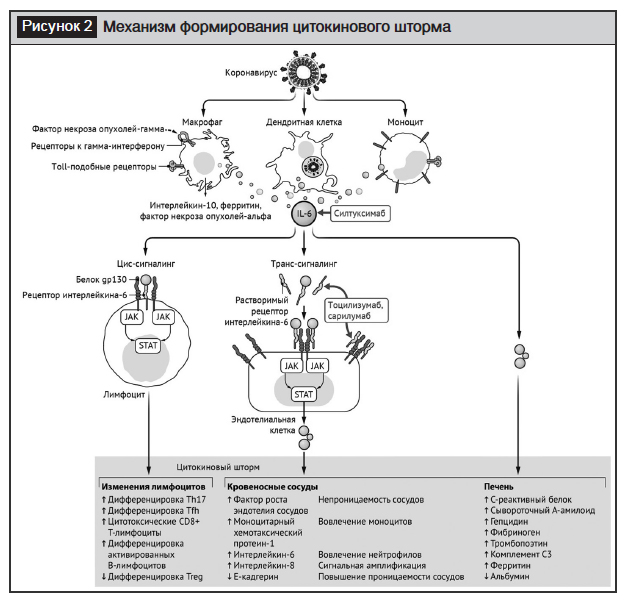
Воспаление развивается в самых разных органах и в сочетании с нехваткой кислорода может привести к их недостаточности. Острый респираторный дистресс-синдром, наблюдаемый при тяжелом течении COVID-19, характеризуется затрудненным дыханием и низким уровнем кислорода в крови. В результате у некоторых пациентов могут развиться вторичные бактериальные и грибковые инфекции. Данный синдром может привести к дыхательной недостаточности, которая является причиной смерти в 70% случаев летальных исходов COVID-19. Кроме того, мощный выброс цитокинов иммунной системой в ответ на вирусную инфекцию и/или вторичные инфекции могут привести к симптомам сепсиса, который является причиной смерти в 28% летальных случаев COVID-19 вследствие неконтролируемого воспаления, приводящего к полиорганной недостаточности, особенно поражая сердечную, печеночную и почечную системы. Большинство пациентов с SARS-CoV-инфекцией, у которых развивалась почечная недостаточность, в конечном итоге умирали. Пока нет окончательного ответа на вопрос, почему у некоторых пациентов COVID-19 протекает так тяжело – отчасти это может быть связано с сопутствующими заболеваниями.
По предварительным данным из разных стран можно сделать вывод, что блокирование гиперактивации иммунитета на уровнях ИЛ-1 и ИЛ-6 может быть эффективно для лечения пациентов с тяжелой формой коронавируса благодаря подавлению или предотвращению цитокинового шторма [9, 34, 46, 50].
Лечение цитокинового шторма, вызванного вирусами, – непростая задача, поскольку он может вызываться разными типами вирусов, отличаться путями передачи инфекции, очагами воздействия инфекции, патогенезом и высвобождаемыми цитокинами.
Помимо поддерживающей терапии [9] лечение цитокинового шторма может осуществляться иммуносупрессией вместе с попытками контроля вызвавшего данное нарушение заболевания или спровоцировавших факторов [14, 46]. Поддерживающее лечение включает в себя интенсивную терапию при нестабильной гемодинамике, поддержку организма при нарушении работы определенных органов и коррекцию коагулопатии, однако сами по себе поддерживающие меры вряд ли будут достаточны для восстановления гомеостаза без воздействия на причины цитокинового шторма [9]. В случае сепсиса, вызванного инфекцией, может оказаться достаточным поддерживающего лечения и противомикробной терапии [50].
Министерство здравоохранения Российской Федерации одобрило проведение клинического исследования двух уже существующих препаратов для лечения тяжелой формы коронавируса. Это олокизумаб (ингибитор ИЛ-6) и RPH-104 (ингибитор ИЛ-1). Рекомендуется определение ИЛ-1, ИЛ-6 вместе с С-реактивным белком для прогноза течения тяжелой формы коронавирусной инфекции. Поскольку для цитокинового шторма наиболее важными цитокинами семейства ИЛ-1 являются ИЛ-1?, ИЛ-18 и ИЛ-33. Наибольший интерес представляют исследования, предметом которых является ингибирование ИЛ-1? с целью снизить проявления цитокинового шторма. [4]. В этой связи рассматривается анакинра, антагонист ИЛ-1?. Он значительно увеличивает коэффициент 28-дневной выживаемости пациентов с тяжелым сепсисом [49]. Клинического опыта применения антагонистов семейства ИЛ-1 для лечения COVID-19 пока нет.
Тоцилизумаб является антигонистом ИЛ-6, супрессирующим функцию иммунной системы. Сегодня тоцилизумаб применяется, главным образом, при аутоиммунных заболеваниях, таких как ревматоидный артрит [7]. Тоцилизумаб сам по себе обладает терапевтическим эффектом при цитокиновом шторме инфекционной природы [7, 40]. У пациентов с тяжело протекающим COVID-19 уровень ИЛ-6 в сыворотке значительно повышен. Клинические исследования, проведенные в Китае, указывают на результативность Тоцилизумаба в лечении пациентов с тяжелыми формами заболевания с обширными двусторонними очагами поражения легких, у которых повышен уровень ИЛ-6 [49].
ФНО – ключевые факторы воспалительного процесса, которые запускают цитокиновый шторм. Они являются важной мишенью для его регулирования. Мета-анализ показал, что анти-ФНО-терапия значительно повышает выживаемость пациентов с сепсисом [53]. Анти-ФНО-терапия достигла также удовлетворительных результатов в лечении неинфекционных заболеваний, таких как атеросклероз [32]. Исследования на модельных животных показали, что ФНО вносит значительный вклад в развитие острого повреждения легких и нарушает Т-клеточный ответ у мышей, зараженных SARS-CoV. У мышей нейтрализация активности ФНО или потеря рецептора ФНО приводит к снижению морбидности и смертности от SARS-CoV [11]. Однако следует отметить, что по крайней мере на поздней стадии инфекции ФНО в сыворотке пациентов с SARS не обнаруживался. Сегодня, несмотря на то, что блокаторы ФНО в терапии пациентов с COVID-19 не рассматривались, возможность их использования заслуживает дальнейших исследований.
Хлорохин ингибирует образование и высвобождение ФНО и ИЛ-6, что указывает на то, что хлорохин может супрессировать цитокиновый шторм у пациентов, инфицированных COVID-19. Хлорохинфосфат использовался в терапии взрослых от 18 до 65 лет в Китае [19, 21].
Таким образом, высокие титры вируса и последующие интенсивные цитокиновый и хемокиновый воспалительные ответы являются причиной повышенной морбидности и смертности, наблюдаемых в патогенезе коронавирусной инфекции HCoV. Опыт лечения SARS и MERS показывает, что снижение вирусной нагрузки посредством медицинского вмешательства на ранних стадиях заболевания и контроль воспалительного процесса с помощью иммуномодуляторов являются эффективными мерами для улучшения прогноза течения коронавирусной инфекции HCoV [5, 16, 20, 36].
Л И Т Е Р А Т У Р А
1. Кетлинский С.А., Симбирцев А.С., Цитокины. – М., 2008. – 552 с.
2. Козлов В.К. Цитокинотерапия: патогенетическая направленность при инфекционных заболеваниях и клиническая эффективность: Рук-во для врачей. – СПб, 2010. – 148 с.
3. Семенов Б.Ф, Зверев В.В. // Журн. микробиол., эпидемиол. и иммунобиол. – 2007. – №4. – С.93–100.
4. Хаитов Р.М., Игнатьева Г.А., Сидорович И.Г. Иммунология. – М., 2002. – 536 с.
5. Arabi Y.M., Shalhoub S., Mandourah Y., et al. // Clin. Infectious Dis.: an official publication of the Infectious Diseases Society of America. 2019. – Vol.70, N9. PubMed PMID: 31925415.
6. Behrens E.M., Koretzky G.A. // Arthritis & Rheumatology. – 2017. – Vol.69, N6. – P.1135–1143.
7. Biggioggero M., Crotti C., Becciolini A., Favalli E.G. // Drug Design, Development Therapy. – 2018. – Vol.13. – P.57–70. PubMed PMID: 30587928.
78. Cameron M.J., Bermejo-Martin J.F., Danesh A., Muller M.P., Kelvin D.J. // Virus Res. – 2008. – Vol.133, N1. – P.13–19. PubMed PMID: 17374415.
9. Canna S.W., Behrens E.M. // Pediatric Clinics North America. – 2012. – Vol.59, N2. – P.329–344.
10. Channappanavar R., Fehr A.R., Zheng J., et al. // J. Clin. Investigation. – 2019. – Vol.130. – P.3625–3639. PubMed PMID: 31355779.
11. Channappanavar R., Fehr A.R., Vijay R., Mack M., et al. // Cell Host & Microbe. – 2016. – Vol.19, N2. – P.181–193. PubMed PMID: 26867177.
12. Chen L., Liu H.-G., Liu W., et al. // Chin. J. Tuberc. Respir. Dis. – 2020. – Vol.43. PubMed PMID: 32026671.
13. Cheung C.Y., Poon L.L.M., Luk W., et al. // J. Virol. – 2005. – Vol.79, N12. – P.7819–7826. PubMed PMID: 15919935.
14. Chousterman B.G., Swirski F.K., Weber G.F. // Sem Immunopathol. – 2017. – Vol.39, N5. – Р.517–528.
15. Clark I.A., Vissel B. // Sem. Immunopathol. – 2017. – Vol.39, N5. – P.505–516.
16. Davidson S., McCabe T.M., Crotta S., et al. // EMBO Molecular Med. – 2016. – Vol.8, N9. – P.1099–1112. PubMed PMID: 27520969.
17. Douda D.N., Jackson R., Grasemann H., Palaniyar N. // J. Immunol. – 2011. – Vol.187, N4. – P.1856–1865. PubMed PMID: 21724991.
18. Drosten C., Seilmaier M., Corman V.M., et al. // Lancet Infectious Dis. – 2013. – Vol.13, N9. – P.745–751. PubMed PMID: 23782859.
19. Expert consensus on chloroquine phosphate for the treatment of novel coronavirus pneumonia / Multicenter collaboration group of Department of Science and Technology of Guangdong Province and Health Commission of Guangdong Province for chloroquine in the treatment of novel coronavirus pneumonia // Chinese J. Tuberculosis Respirat. Dis. – 2020. – Vol.43, N3. – P.185–188. PubMed PMID: 32075365.
20. Falzarano D., de Wit E., Rasmussen A.L., et al. // Nature Med. – 2013. – Vol.19, N10. – P.1313–1317. PubMed PMID: 24013700.
21. Gao J., Tian Z., Yang X. // Bio Science Trends. – 2020. – Vol.14, N1. – P.72–73. PubMed PMID: 32074550.
22. García-Sastre A., Biron C.A. // Science. – 2006. – Vol.312 (5775). – P.879–882. PubMed PMID: 16690858.
23. Herold S., Steinmueller M., von Wulffen W., et al. // J. Experiment. Med. – 2008. – Vol.205, N13. – P.3065–3077. PubMed PMID: 19064696.
24. Högner K., Wolff T., Pleschka S., et al. // PLoSpathogens. – 2013. – Vol.9, N2. – P.1003188. PubMed PMID: 23468627.
25. Huang C., Wang Y., Li X., et al. // Lancet. – 2020. – Vol.395 (10223). – P.497–506.
26. Jiang Y., Xu J., Zhou C., et al // Am. J. Resp. Crit. Care Med. – 2005. – Vol.171, N8. – P.850–857. PubMed PMID: 15657466.
27. Kim E.S., Choe P.G., Park W.B., et al. // J. Korean Med. Sci. – 2016. – Vol.31, N11. – P.1717–1725. PubMed PMID: 27709848.
28. Kuiken T., Fouchier R.A.M., Schutten M., et al. // Lancet. – 2003. – Vol.362 (9380). – P.263–270. PubMed PMID: 12892955.
29. Lau S.K.P., Lau C.C.Y., Chan K.-H., et al. // J. Gen. Virolo. – 2013. – Vol.94, N12. – P.2679–2690. PubMed PMID: 24077366.
30. Law H.K.W., Cheung C.Y., Ng H.Y., et al. // Blood. – 2005. – Vol.106, N7. – P.2366–2374. PubMed PMID: 15860669.
31. Marchingo J.M., Sinclair L.V., Howden A.J.M., Cantrell D.A. // eLife. – 2020. – Vol.9. – e53725. PubMed PMID: 32022686.
32. McDermott J.E., Mitchell H.D., Gralinski L.E., et al. // BMC Systems Biology. – 2016. – Vol.10, N1. – P.93. PubMed PMID: 27663205.
33. Min C.K., Cheon S., Ha N.Y., et al. // Scientific Rep. – 2016. – Vol.6. – P.25359. PubMed PMID: 27146253.
34. Moore J.B., June C.H. // Science. – 2020. – Vol.368 (6490). – P.473–474.
35. Ng D.L., Al Hosani F., Keating M.K., et al. // Am. J. Pathol. – 2016. – Vol.186, N3. – P.652–658. PubMed PMID: 26857507.
36. Omrani A.S., Saad M.M., Baig K., et al. // Lancet Infectious Dis. – 2014. – Vol.14, N11. – P.1090–1095. PubMed PMID: 25278221.
37. Parsons P.E., Eisner M.D., Thompson B.T., et al. // Crit. Care Med. – 2005. – Vol.33, N1. – P.1–232. PubMed PMID: 15644641.
38. Peck K.M., Burch C.L., Heise M.T., Baric R.S. // Ann. Rev. Virol. – 2015. – Vol.2, N1. – Р.95–117. PubMed PMID: 26958908.
39. Peiris J.S.M., Lai S.T., Poon L.L.M., et al. // Lancet. – 2003. – Vol.361 (9366). – P.1319–1325. PubMed PMID: 12711465.
40. Qiu P., Cui X., Sun J., et al. // Critical Care Medicine. – 2013. – Vol.41, N10. – P.2419–2429. PubMed PMID: 23887234.
41. Ranieri V.M., Rubenfeld G.D., Thompson B.T., Ferguson N.D., et al. // JAMA. – 2012. – Vol.307, N23. – P.2526–2533.
42. Reghunathan R., Jayapal M., Hsu L.Y., et al. // BMC Immunol. – 2005. – Vol.6. – P.2. PubMed PMID: 15655079.
43. Rockx B., Baas T., Zornetzer G.A., et al. // J. Virol. – 2009. – Vol.83, N14. – P.7062–7074. PubMed PMID: 19420084.
44. Rodrigue-Gervais I.G., Labbé K., Dagenais M., et al. // Cellhost & Microbe. – 2014. – Vol.15, N1. – P.23–35. PubMed PMID: 24439895.
45. Scheuplein V.A., Seifried J., Malczyk A.H., et al. // J. Virol. – 2015. – Vol.89, N7. – P.3859–3569. PubMed PMID: 25609809.
46. Shimizu M. Clinical Features of Cytokine Storm Syndrome // Springer Nature. – 2019. – P.31–34.
47. Smits S.L., de Lang A., van den Brand J.M.A., et al. // PLoSpathogens. – 2010. – Vol.6, N2. – P.e1000756. PubMed PMID: 20140198.
48. Stockman L.J., Bellamy R., Garner P. // PLoSmedicine. – 2006. – Vol.3 (9). – e343. PubMed PMID: 16968120.
49. Tanaka T., Narazaki M., Kishimoto T. // Immunotherapy. – 2016. – Vol.8, N8. – P.959–970. PubMed PMID: 27381687.
50. Tay M.Z., Poh C.M., Rénia L., et al. // Nature Rev. Immunol. – 2020. – Vol.20, N6. – P.363–374.
51. Teijaro J.R. // Sem. Immunopathol. – 2017. – Vol.39, N5. – P.501–503.
52. Tynell J., Westenius V., Rönkkö E., et al. // J. Gen. Virol. – 2016. – Vol.97, N2. – P.344–355. PubMed PMID: 26602089.
53. Udalova I., Monaco C., Nanchahal J., Feldmann M. // Microbiol. Spectrum. – 2016. – Vol.4, N4. – P.1–11. PMID: 27726761.
54. Wang H., Ma S. // Am. J. Emerg. Med. – 2008. – Vol.26, N6. – P.711–715. PubMed PMID: 18606328.
55. Weiss S.R., Navas-Martin S. // Microbiol. Mol. Biol. Rev. – 2005. – Vol.69, N4. – P.635–664. PubMed PMID: 16339739.
56. Yang X., Yu Y., Xu J., et al. // Lancet Resp. Med. – 2020. – Vol.8, N5. – P.475–481. PubMed PMID: 32105632.
57. Zhou J., Chu H., Li C., et al. // J. Infect. Dis. – 2014. – Vol.209, N9. – P.1331–1342. PubMed PMID: 24065148.
Медицинские новости. – 2020. – №10. – С. 4-8.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.