Диабетическая полинейропатия — самое частое и грозное осложнение сахарного диабета (СД). Распространенность ее колеблется в достаточно широких пределах (от 15 до 90%), причем у лиц с длительностью СД 15 лет и более это осложнение регистрируется более чем в 60% случаев. При манифестации СД 2 типа от 2 до 6% пациентов уже имеют проявления нейропатии. Распространенность автономной нейропатии, независимо от типа СД, составляет примерно 25–35% [5, 12, 13, 17].
Несмотря на совершенство современных методов диагностики, нейропатия часто остается нераспознанной на начальных стадиях развития, поскольку в течение некоторого периода времени она развивается субклинически и пациенты, как правило, не предъявляют специфических жалоб. В результате в клинической практике недооцениваются ранние стадии развития нейропатии, медикаментозная помощь на которых максимально эффективна. Симптоматическая терапия не может полностью удовлетворить клиницистов, так как она не устраняет основные причины метаболических нарушений и сопровождается побочными эффектами.
Цель классической терапии диабетической нейропатии (ДН) — достижение и поддержание постоянной стойкой нормогликемии и применение препаратов патогенетического действия [4, 12]. Именно такой подход, по данным многочисленных проспективных исследований, является оптимальным, и одно из ведущих мест в терапевтической практике занимают препараты витаминов группы В.
Этиопатогенетические основы терапевтической эффективности препаратов тиамина
В основе патогенеза нейропатии лежат две взаимодополняющие теории: метаболическая и микрососудистая, согласно которым все изменения индуцируются хронической гипергликемией.
Основные положения метаболической теории:
1. Активация полиолового пути обмена глюкозы, что сопровождается значительным повышением синтеза сорбитола под действием альдозоредуктазы и накоплением образующейся из него фруктозы, не способных преодолеть клеточную мембрану. В результате происходит повреждение клеток за счет внутриклеточной гиперосмолярности и связанной с ней гипергидратации.
2. Снижение образования оксида азота (оказывает выраженное релаксирующее действие на сосуды), являющегося следствием увеличения в мембранах и клетках уровня свободных радикалов, активации протеинкиназы С и снижения образования НАДФ, участвующего в синтезе оксида азота.
На фоне этих процессов происходит:
• снижение концентрации миоинозитола (энергетический источник нейронов), в результате чего развивается демиелинизация нервных волокон и снижается скорость проведения нервного импульса;
• снижение Na-K-AТФ-азной активности (также приводит к демиелинизации нервных волокон и снижению передачи нервного импульса по волокну);
• изменение метаболизма фосфоинозитида (энергетический источник нейронов), что также ведет к снижению скорости проведения нервного импульса;
• неферментное гликирование белков, нарушение синтеза липидов в миелиновой оболочке и поступления витаминов при СД могут в комплексе приводить к демиелинизации и снижению интраневрального кровотока;
• нарушение образования факторов роста (защищают нейроны от поражения и способствуют их регенерации и увеличению плотности нервных волокон; недостаток факторов роста ведет к аксонопатии) [1, 16].
В результате из-за нарушения гексозаминового пути метаболизма глюкозы происходит накопление конечных продуктов гликирования (КПГ; в англоязычной литературе — AGEs (advanced glycation endproducts)).
В основе микрососудистой теории лежит гипотеза гипоксии-ишемии нервной ткани, вследствие чего развивается микроангиопатия vasa nervorum и происходит ряд реологических изменений.
КПГ индуцируют развитие нейропатии путем:
- индукции микроангиопатии аксональных сосудов (vasa nervorum);
- гликирования протеинов миелиновой оболочки;
- гликирования внутриаксональных протеинов;
- взаимодействия с КПГ-рецепторами на нервных и глиальных клетках.
Поражение тонких и толстых нервных волокон при ДН способствует снижению всех видов периферической чувствительности, ослаблению периферических сухожильных рефлексов, что влечет за собой изменения анатомо-функционального строения стопы, обусловливая инициацию процесса язвообразования с развитием в последующем гангрены и ампутацией [5].
Энергетическим источником для нервных волокон является результат метаболизма глюкозы в цикле трикарбоновых кислот (ЦТК, цикл Кребса), происходящего с высвобождением энергии. ЦТК имеет два дегидрогеназных комплекса, замедление функции которых может привести к остановке деятельности всего цикла. Эти комплексы представлены одним кофактором (ионы магния), тремя ферментами и пятью коферментами, главные из которых — тиаминпирофосфат (В1) и α–липоевая кислота. Дефицит этих коферментов способствует развитию нейропатии. Витамин В1 (тиамин) принимает активное участие во многих этапах метаболизма нервной ткани, в частности в пентозофосфатном пути окисления глюкозы (рис. 1).
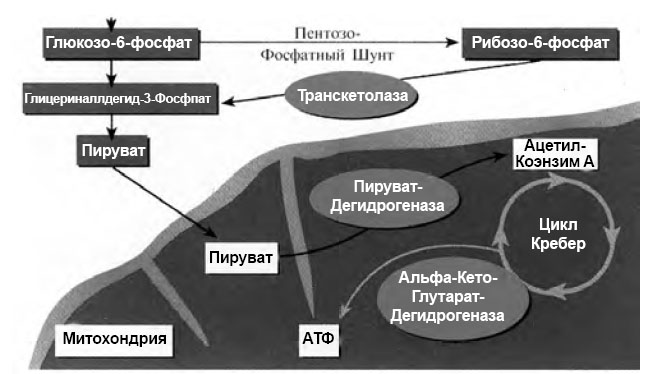
Рис. 1. Тиамин в качестве кофермента в углеводном обмене (по Bernstein, 2005)
При поступлении тиамина в клетку активность транскетолазы, ослабленная при СД, возрастает в несколько раз. Кроме того, токсичность глюкозы уменьшается вследствие реакции введения глицеринальдегид–3–фосфата в пентозофосфатный шунт и пирувата в ЦТК; благодаря «сбросу» на пируват снижается концентрация промежуточных метаболитов и предотвращается образование КПГ (рис.1). Фосфоэфир тиамина и сам тиамин модулируют передаточную функцию ацетилхолина. Тиаминпирофосфат включается в натриевые нейрональные каналы нервных стволов и предположительно принимает участие в контроле за скоростью проведения возбуждения по нервному волокну. Тиамин вмешивается в ноцицептивные процессы в спинном мозге и уменьшает болевые ощущения, на чем основано его аналгетическое действие.
При дефиците тиамина нарушается синтез оксида азота, что приводит к вазоспазму и нарушениям вегетативной нервной системы [7].
Таким образом, применение тиамина и антиоксидантов для терапии ДН патогенетически оправдано. Антиоксиданты снижают выраженность оксидативного стресса, улавливая повреждающие нервные клетки свободные радикалы, а тиамин увеличивает активность транскетолазы, обеспечивая утилизацию промежуточных продуктов гликолиза в пентозофосфатном цикле.
Клиническая эффективность бенфотиамина в терапии диабетической нейропатии
Ограничением для применения водорастворимой формы тиамина внутрь в терапевтических дозах является его относительно низкая биодоступность, поскольку водорастворимый тиамин плохо всасывается и частично разрушается тиаминазой кишечника, в котором он в основном абсорбируется. Решением этой проблемы стало создание жирорастворимых форм тиамина, не имеющих недостатков, свойственных водорастворимой пероральной форме этого витамина. Такие формы были синтезированы в Японии в 50–х годах ХХ в., и вся группа жирорастворимых тиаминов была названа аллитиаминами. Среди них наибольшей биодоступностью и способностью проникать в клетку обладает бенфотиамин.
Липофильная форма тиамина выгодно отличается от гидрофильной по нескольким причинам. Липофильные формы (бенфотиамин) характеризуются более высокой биодоступностью, ускоренной резорбцией и более быстрым периодом насыщения (рис. 2). Как показали K. Schreeb et al., уровень тиамина в плазме примерно в 5–6 раз выше при введении бенфотиамина, чем после использования водорастворимой формы препарата [10, 11]. Абсолютная биодоступность по прошествии недели была в 3,6 раза выше. Среди липофильных производных витамина В1 бенфотиамин имеет самую высокую биодоступность.
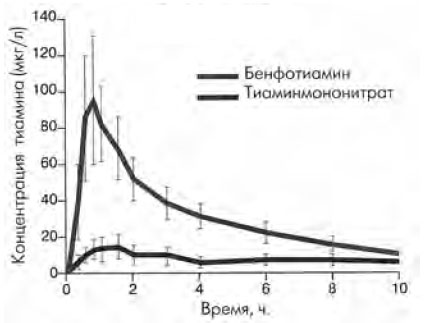
Рис. 2. Сравнительная характеристика концентраций бенфотиамина и тиаминмононитрата в плазме крови (K.H. Schreeb, 1997)
Как показано в контролированных исследованиях [2], именно с концентрацией бенфотиамина в крови связана его клиническая эффективность. Биодоступность гидрофильного тиамина при приеме внутрь ограничивается его разрушением в ЖКТ под воздействием тиаминазы и ограничением всасывания, не зависящего от дозы назначаемого препарата. После повышения концентрации тиамина в крови более 2 мкмоль/л активный натрийзависимый транспорт его сменяется на менее эффективную пассивную диффузию. Максимально возможная абсорбция из ЖКТ составляет 10%, а за сутки способно всосаться не более 15 мг. При высоких концентрациях в крови тиамин активно выводится почками практически в неизмененном виде. Липофильный бенфотиамин в ЖКТ превращается в тиамин, частично фосфорилированный, и уже в таком виде всасывается в кровь и распределяется по организму. Кроме того, при абсорбции его в ЖКТ отсутствует эффект насыщения, и поступление в кровь определяется его концентрацией в просвете кишечника.
Среди имеющихся в настоящее время на белорусском фармацевтическом рынке комбинированных препаратов, содержащих витамины группы В, только Мильгамма® драже (Вёрваг Фарма ГмбХ, Германия) содержит жирорастворимый бенфотиамин. Мильгамма® драже состоит из 100 мг бенфотиамина и 100 мг пиридоксина.
В многочисленных исследованиях доказана клиническая эффективность бенфотиамина при ДН [2, 9, 10]. В табл. 1 приведена сравнительная характеристика проведенных клинических исследований.
Таблица 1. Сравнительная характеристика клинических исследований по эффективности бенфотиамина
|
|
|
|
|
|
|
|
|
|
|
Рандомизированное двойное слепое плацебо-контролированнное (pilot)
|
|
Бенфотиамин 300 мг/сут в течение 3 нед.
|
|
|
|
|
|
|
Бенфотиамин 400 мг/сут в течение 3 нед., затем 300 мг/сут в течение 8 нед.
|
|
|
|
|
Рандомизированное двойное слепое плацебо–контролированнное (pilot)
|
|
Бенфотиамин 240 мг/сут в течение 2 нед., затем 120 мг/сут в течение 10 нед.
|
|
|
|
|
Рандомизированное плацебо–контролированное
|
|
Мильгамма® 1 драже 3 раза в день в течение 6 нед.
|
|
|
|
|
Рандомизированнное, двойное слепое, плацебо–контролированнное (pilot)
|
|
Бенфотиамин 400 мг/сут в течение 3 нед.
|
В исследовании О.А. Маркиной (сравнивались три группы пациентов: получавшие Мильгамму® по 1 драже 3 раза в день, водорастворимые соли витаминов В1 и В6 по 100 мг в/м и плацебо) показано, что на фоне парентерального введения тиамина (водорастворимая форма) происходило более быстрое повышение концентрации тиамина в плазме и гемолизате, но, начиная с 14-го дня лечения, концентрация тиамина в плазме на фоне приема Мильгаммы® (1 драже 3 раза в день в течение 6 нед.) достоверно превышала таковую в группе больных, получавших водорастворимый тиамин, и оставалась на этом уровне до конца лечения [2]. У всех пациентов, получавших Мильгамму® драже, отмечалось снижение выраженности неврологических проявлений. В недавнем исследовании [9] продемонстрировано достоверное уменьшение всей симптоматики при нейропатии (болей, судорог и дизестезий, рис. 3) по показателям шкалы нейропатического дисфункционального счета (НДС).
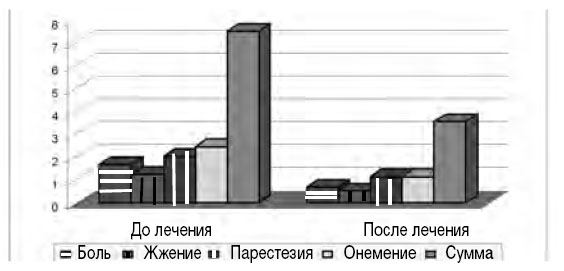
Рис. 3. Положительная динамика симптомов диабетической нейропатии по шкале НДС в результате применения бенфотиамина в дозе 300 мг/сут (BEDIP study group, 2005; Р< 0,005–0,01)
Таким образом, по результатам клинических исследований убедительно доказано положительное терапевтическое действие бенфотиамина при диабетической дистальной полинейропатии (ДПН), улучшение функции соматических двигательных и чувствительных нервов и вегетативных нервных волокон.
Весьма интересны в клиническом плане научные сообщения о лечении бенфотиамином других осложнений СД. Имеются данные, подтверждающие клиническую эффективность бенфотиамина при синдроме диабетической стопы (СДС), как известно, часто приводящем к ампутации нижних конечностей. В 85% случаев этим ампутациям предшествуют трофические язвы стоп, а одной из причин развития язвенных дефектов является ДПН. В исследовании, проведенном на базе ЭНЦ РАМН [3], показано, что на фоне приема Мильгаммы® (1 драже 3 раза в день в течение 6 нед.) отмечено достоверно большее количество пациентов с заживлением язвенных дефектов к концу исследования или выраженное уменьшение размеров трофических язв (рис. 4).
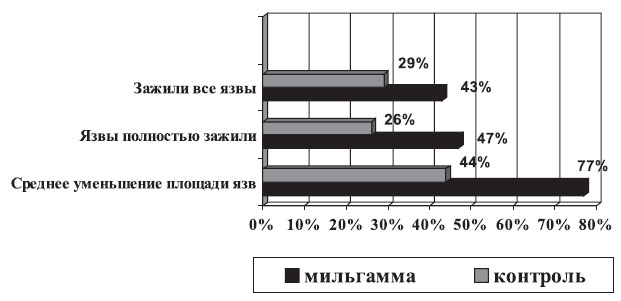
Рис. 4. Эффективность Мильгаммы в лечении СДС [3]
В экспериментах, проведенных на крысах, американскими учеными показано, что бенфотиамин защищает сетчатку глаза при диабетической ретинопатии, повышая содержание транскетолазы на 400% и снижая содержание КПГ, при этом восстанавливались протеинкиназа С и фактор транскрипции NF–кappaB [8]. В недавнем экспериментальном исследовании установлена защитная роль бенфотиамина в развитии диабетической нефропатии; на фоне приема этого препарата получены данные, свидетельствующие об отдалении периода начала гиперфильтрации и потери альбумина с мочой на 76% [14, 15].
Кроме того, имеются исследования, посвященные изучению превентивного действия бенфотиамина при автономной нейропатии. В исследовании Z. Koltai [6] при аллоксан–индуцированном диабете у собак показано, что назначение 50 мг бенфотиамина в сутки в течение 3 мес не приводит к развитию автономной нейропатии сердца в основной группе по сравнению с контрольной (рис. 5).
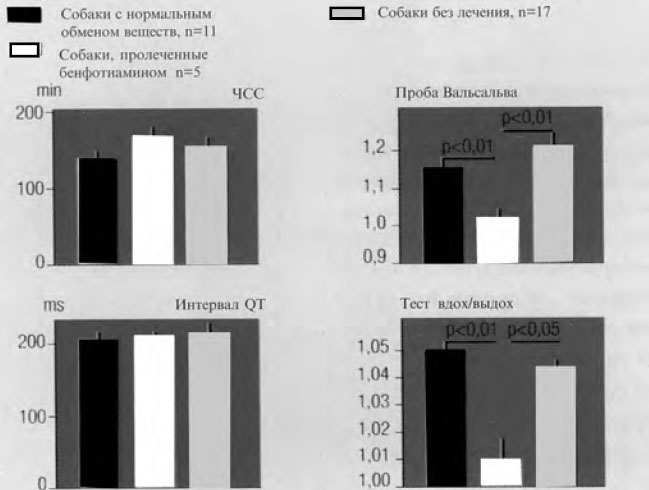
Рис. 5. Автономные параметры при экспериментальном диабете с лечением и без лечения бенфотиамином (по Z. Koltai)
Итак, полученные к настоящему времени научные данные по использованию бенфотиамина в терапии осложнений сахарного диабета свидетельствуют о его ведущей патогенетической роли в лечении диабетической нейропатии и возможном успешном применении при других осложнениях диабета, приводящих к стойкой потере трудоспособности и инвалидности. В США бенфотиамин проходит стадии экспериментальных исследований и клинических испытаний, причем подчеркивается его «абсолютный потенциал» в лечении и предупреждении развития таких грозных последствий СД, как слепота, почечная недостаточность, поражения эндотелия сосудов, ведущие к инфарктам и инсультам. В связи с этим многие специалисты предсказывают бенфотиамину большое будущее в профилактике отдаленных последствий сахарного диабета.
1. Балаболкин М.И. // Сахарный диабет. — М., 1994. — Гл. 5. — С. 224—236.
2. Маркина О.А. // Клин. фармакология и терапия. —2003.— № 2.— С. 6–9.
3. Удовиченко О.В., Курцева Т.Г. // Сахарный диабет.— 1999.— № 2 (3). — С. 4–16.
4. Boulton A.J.M. // Gac. Med. Mex. — 1994. — Vol. 130, N 1. — P. 18–25.
5. Dyck P.J., Dyck P.J.B. // Diabetic Neuropathy / eds.: P.J. Dyck, P.K. Thomas; 2nd ed.—Philadelphia: W.B. Saunders, 1999. — P. 255–278.
6. Greis F.A. // Полинейропатии в диалогах. — 2003.— № 1. — С. 5–6.
7. GröberU. // Deutscher. Apoteker. Zeitung. — 2005. — Vol. 37. — P.122–126.
8. Hammes H.P., Du X., Edelstein D. et al. // Nat. Med.— 2003. — Vol. 9(3). — P. 294–299.
9. Haupt E., Ledermann H., Köpcke W. // Clin. Pharmacol. & Ther. — 2005. — Vol. 43, N 2.— P. 71–77.
10. Schreeb K.H., Freudenthaler S., Vormfelde S.V. et al. // Eur. J. Clin. Pharmacol. — 1997. — Vol. 52. — P.315–320.
11. Stirban A., Negrean M., Stratmann B. et al. // Diabetes Care. — 2006. — Vol. 29, N 9. — P. 2064–2071.
12. The DCCT Research Group. The effect of intensive diabetes therapy on the development and progression of neuropathy // Ann. Intern. Med. — 1995.—Vol. 122. — P. 561–568.
13. The epidemiology of diabetic neuropathy. DiaCAN Multicenter Study Group / D. Ziegler, F.A. Gries, M. Spuler, F. Lessmann. — Diabetes Research Institute, Heinrich-Heine-University, Dusseldorf, Germany // Diabet. Med. — 1993.— Vol. 10, suppl. 2. — P. 82S—86S.
14. Thornalley P.J., Babaei-Jadidi R., Al Ali H. et al. // Diabetologia. — 2007. — Vol. 50 (10). — P.2164 – 2170.
15. Yao D., Taguchi T., Matsumura T. et al. // J. of Biol. Chem. — 2007. — Vol. 282 (42). — P.31038 – 31045.
16. Yorck M.A., Dunlap J.A., Ginsberg B.N. // J. Neurochem. – 1987. – Vol. 48. – P. 53—61.
17. Ziegler D. // Diabet. Metab. Rev. — 2003. — Vol. 10. — P.339–383.
Медицинские новости. – 2008. – №3. – С. 41-44.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.