На сегодняшний день актуальность изучения проблемы сахарного диабета II типа (СД II) в детской популяции не вызывает сомнения [5, 9, 25, 29, 39, 41]. Число больных этим заболеванием в разных странах составляет 8–45% общего количества детей с диабетом [7, 9, 10, 29]. За последнее время в нашей республике распространенность этой эндокринной патологии среди детского населения увеличилась более чем на 60%.
СД II является гетерогенным заболеванием, развивающимся в результате сочетания врожденных и приобретенных факторов [4]. Генетическая составляющая этой эндокринопатии отражена в табл. 1 [29]. Первичный генетический дефект, лежащий в ее основе, полностью не раскрыт. Выявленные редкие генетические мутации составляют менее 5% случаев СД II [29]. Очевидно, развитие заболевания определяет совокупность множества генов, контролирующих как синтез инсулина островковыми клетками, так и его взаимодействие с клетками-мишенями в периферических органах [2].
|
Факторы, связанные с инсулином
|
Факторы, секретируемые жировой тканью
|
|
|
|
Субстраты инсулинового рецептора (IRS-1–IRS-4)
|
|
|
|
|
Энзимы (фосфатазы, серинкиназы, тирозинкиназы, фосфатидилинозитол-3-киназа)
|
|
|
|
Специфические тканевые факторы
|
|
Транспортеры глюкозы (GLUT4)
|
|
|
|
|
Энзимы (фосфоенолпируват карбоксикиназа)
|
|
|
|
|
Рецепторы сульфонилмочевины
|
Все компоненты ренин-ангиотензиновой системы
|
Инсулиноподобный фактор роста -1, -2
|
|
|
Таблица 1. Гены-кандидаты, участвующие в регуляции гомеостаза глюкозы у человека и патогенезе СД II [29]
СД II у детей – сложная многофакторная и полигенная патология, возникающая при тесном взаимодействии генетических и средовых компонентов. Ранней манифестации заболевания в детской популяции способствуют избыточная масса тела и ожирение, низкая физическая активность, употребление высококалорийных продуктов питания, частая распространенность начальных патологических изменений углеводного обмена, масса тела при рождении менее 2500 г, физиологическая пубертатная инсулинорезистентность (ИР), стрессовые состояния, урбанизация, определенная этническая принадлежность [5, 8, 11, 12, 15, 20, 23, 25, 27–30, 32, 36].
Спектр клинических проявлений при постановке диагноза у детей варьирует от бессимптомной гипергликемии до диабетического кетоацидоза и гипергликемического гиперосмолярного состояния [5, 6]. При манифестации СД II у детей сформировано двойное нарушение: сочетание дисфункции β-клеток и сниженной чувствительности периферических тканей к инсулину (инсулинорезистентность) (табл. 2) [29]. Дисфункция β-клеток проявляется изменением синтеза или превращения инсулина, нарушением его секреции [4]. Периферическая инсулинорезистентность выражена относительно и длительное время компенсируется избыточной продукцией инсулина β-клетками (гиперинсулинемией), поддерживая углеводный обмен в норме. Гиперинсулинемия приравнивается к косвенным признакам ИР и рассматривается в качестве предвестника развития СД II у детей [3, 4, 22].
Чувствительность к инсулину
|
Нарушение инсулиновой чувствительности
|
|
|
Секреция инсулина (гипо-, гиперинсулинемия)
|
|
|
Клиренс инсулина
(Контр)регуляторный ответ (резистин, глюкагон, лептин)
|
Таблица 2. Факторы, способствующие нарушению гомеостаза глюкозы при развитии и прогрессировании СД II [29]
Наиболее клинически значимым при прогрессировании СД II является снижение инсулиновой чувствительности мышечной, жировой и печеночной тканей [42]. ИР мышечной ткани характеризуется уменьшением поступления глюкозы из крови в миоциты и ее утилизации в них. ИР жирового депо выражается в уменьшении чувствительности к антилиполитическому действию инсулина, ведущему к накоплению глицерина и свободных жирных кислот – источнику образования атерогенных липопротеидов. ИР ткани печени проявляется понижением синтеза гликогена и активацией процессов гликогенолиза и глюконеогенеза. В процессе развития СД II, при начинающемся истощении секреторной функции β-клеток и относительном снижении гиперинсулинемии, вначале страдает функция захвата глюкозы мышечной тканью, затем – гликогенсинтетическая способность печени и в последнюю очередь – снижение липолитической функции жировой ткани [4, 22].
Установлена генетическая предрасположенность к развитию ИР [4, 9]. Клинически она реализуется (в виде метаболического синдрома и (или) СД II) только при влиянии на организм ребенка совокупности соответствующих внешних факторов: избыточного высококалорийного питания, низкой физической нагрузки, индекса массы тела более 85 перцентили для данного пола и возраста, начале полового созревания, принадлежности к определенной расе/этнической группе и др. [25].
Основные метаболические проблемы при СД II разделяют на обратимые и необратимые. Говоря о ИР, к необратимым относятся генетически обусловленные, прогрессирующие рецепторные и пострецепторные дефекты на уровне печени, жировой и мышечной тканей, применительно к дефектам секреции инсулина – генетически обусловленные, прогрессирующие нарушения, способствующие снижению массы β-клеток, связанные с возрастом, наряду с деструкцией β-клеток и дефектами регенерации [4]. К обратимым изменениям при ИР относят глюкозотоксичность и липотоксичность [4].
Роль ранней манифестации у детей СД II и ИР заключается в развитии и прогрессировании сердечно-сосудистой патологии и дислипидемии [3, 18, 20, 29, 32, 34]. Это определяет актуальность изучения современных подходов к коррекции гипергликемии и феномена ИР при СД II у детей.
Цель нашего исследования – выявление клинических и метаболических особенностей проявления СД II у детей и оценка адекватности схем проводимой сахароснижающей терапии.
Материалы и методы
В исследование включено 20 больных (м/д=9/11) СД II в возрасте 12,2 –16,9 года (средний возраст – 14,9 ± 1,6 года), находившихся на лечении в Республиканском детском эндокринологическом центре в 2007–2008 гг. У всех пациентов собран анамнез с уточнением срока гестации, роста и массы тела при рождении, наличия нарушений углеводного и липидного обмена у родственников, рассчитан и оценен суточный калораж пациента относительно его фактических потребностей, определен уровень физической активности.
Оценку антропометрических параметров проводили с использованием индексов массы тела (ИМТ), соотношения окружности талии к окружности бедер (ОТ/ОБ). Показатели ИМТ более 97 перцентили для данного возраста и пола рассматривали как ожирение [15, 17, 41, 43]. Величины ОТ/ОБ более 0,85 у девочек и 0,9 у мальчиков указывали на абдоминальную форму ожирения. Уровни артериального давления, превышавшие при трехкратном измерении 97 перцентиль для пола и возраста, относили к повышенным [1].
Чувствительность тканей к инсулину оценивали по косвенным показателям: базальным уровням инсулинемии и гликемии, расчетным индексам (инсулин базальный/гликемия базальная (И/Г)), малой модели гомеостаза с определением параметра HOMAIR (Homeostatic Model Assessment) [35, 45]. Согласно рекомендациям Американской кардиологической ассоциации (АНА, 2003), у детей базальные значения инсулинемии в норме не превышают 15 мМЕ/л. Интервал значений 15–20 мМЕ/л характерен для пограничной гиперинсулинемии, более 20 мМЕ/л – для высокой [3]. Показатели HOMAIR более 2,77 свидетельствовали о ИР [3, 4]. Степень компенсации углеводного обмена оценивали по уровням гликированного белка – фруктозамина (ФА) (норма до 285 мкмоль/л). Концентрацию общего холестерина (ОХ), триглицеридов (ТГ) определяли ферментативным, гликемию – ферментативным глюкозооксидантным методами с помощью наборов реагентов «Cormay». В качестве критериев оценки холестеролемии применяли рекомендации Национальной образовательной программы по холестерину для детей и подростков, согласно которым уровни более 5,2 ммоль/л считались высокими [1, 3]. Радиоиммунным методом изучали в сыворотке показатели инсулина (наборы ХОП ИБОХ НАНБ) и С-пептида (наборы «Immunotech», Чехия). Определяли радиоиммунным методом уровни антител к глютаматдегидрогеназе (GAD 65) (норма < 1,0 Ед/мл) (наборы «Immunotech»).
Обработку полученных результатов проводили при помощи пакета статистических программ Statistica 6.0, различия считали достоверными при P < 0,05.
Результаты и обсуждение
Для обследованных больных СД II характерен отягощенный семейный анамнез по ожирению (65%). ИМТ более 30 кг/м2 отмечался у обоих родителей в 25%, у одного – в 40% случаев. Более половины детей (55%) имели родственников, страдающих ожирением. Ожирение у родных сибсов наблюдалось у 2 больных. Установлен высокий процент (60%) распространенности СД II среди родственников 1 и 2 степени родства (по материнской линии – у 75% пациентов, по отцовской – у 25%), подтверждающий наследственный характер нарушений углеводного обмена [25].
По мнению ряда авторов, предрасположенность к развитию ИР и СД II у детей развивается внутриутробно [14, 30, 31, 33]. Постнатальное формирование гиперинсулинемии и ИР является отсроченными последствиями задержки внутриутробного развития и макросомии. Макросомия плода рассматривается как результат действия генетических механизмов, направленных на регуляцию секреции фетального инсулина и определяющих чувствительность к нему фетальных тканей [3]. Ее наличие указывает на существовавшую эмбриональную гиперинсулинемию [3].
Согласно гипотезе о «бережливом генотипе», разработанной J.V. Neel [33], развитие ИР представляет компенсаторно-приспособительную реакцию на алиментарный дефицит нутриентов. Ее значение заключается в адаптации и выживании эмбриона при патологическом течении беременности [3, 31]. Модель эмбриональной ИР сохраняется в генетической памяти ребенка и продолжает работать постнатально на накопление энергии при избыточном поступлении калорий. Это приводит к раннему ожирению, нарушению углеводного обмена, дислипидемии в детском возрасте [25]. Задержка внутриутробного развития усиливает апоптоз β-клеток поджелудочной железы, снижая их массу. Уменьшение массы β -клеток является триггером ранней манифестации СД II у детей при наличии генетической предрасположенности и влиянии внешнесредовых диабетогенных факторов [4, 22, 25]. Таким образом, для развития СД II прогностически неблагоприятным является рождение маловесных детей к сроку гестации [3, 30]. Проведенный нами анализ антропометрических показателей при рождении показал соответствие средних значений массы тела (3550 ± 150 г) и роста (54 ± 2 см) больных СД II популяционным нормам. Не выявлено случаев рождения маловесных и крупновесных к нормальному сроку гестации детей.
Одним из значимых факторов, участвующих в генезе ИР при СД II у детей, является процесс полового созревания. Согласно результатам нашего исследования, 80% детей с СД II были пубертатного возраста и 20% – допубертатного. Для пубертата характерны физиологическая гиперинсулинемия, снижение чувствительности к инсулину и формирование относительной ИР [8]. Развитие ИР сопровождается активацией ростовой оси: повышается концентрация соматотропного гормона и инсулиноподобных факторов роста, снижается уровень связывающего их белка [3].
Избыточная масса тела в подростковом возрасте также способствует усилению физиологической гиперинсулинемии и ИР [3]. В работе M. Wabitsch выявлен высокий процент сопутствующих повышенных сывороточных уровней инсулина (67%) и ИР (индекс HOMAIR более 3,8) (79%) у детей с ожирением [44]. Величины базальной гиперинсулинемии и ИР возрастали пропорционально степени ожирения. По мнению некоторых авторов, ожирение в детском возрасте, особенно по абдоминальному типу, является фактором риска ранней манифестации СД II [3, 18, 20, 27–29, 32, 34].
У 10% обследованных пациентов зарегистрировано повышение показателей ИМТ > 90 перцентили, у 45% > 97 перцентили (из них 78% – девочки). Величины индекса ОТ/ОБ у больных СД II на фоне ожирения соответствовали абдоминальному типу отложения жира (мальчики – 0,92 ± 0,04, девочки – 0,85 ± 0,02), что указывало на повышенный риск формирования других метаболических нарушений (артериальной гипертензии, гиперурикемии, дислипидемии и т.д.). Низкий уровень (35%) выявленного нами акантозиса нигриканса как одного из клинических маркеров ИР и СД II [25] может быть обусловлен небольшой выборкой больных. У 6 детей отмечено повышение артериального давления (систолическое/диастолическое 135,37±3,1/85,60±1,4 мм рт. ст.). Нами не установлено увеличения уровней ОХ (4,52 ± 0,26 ммоль/л) и ТГ (1,27 ± 0,38 ммоль/л) в группе обследованных.
Уровни тощаковой гликемии при первичном обращении колебались от 5,8 до 11,8 ммоль/л (6,6 ± 1,8 ммоль/л). Средние значения базального С-пептида составили 829,8 ± 317,8 пмоль/л и входили в пределы нормы диагностического набора (160–1100 пмоль/л), что свидетельствовало о сохранении функции β-клеток поджелудочной железы у всех больных. Уровни антител к GAD 65 соответствовали 0,61 ± 0,32 Ед/мл. Случаев кетонурии не зарегистрировано.
Исходные показатели базального инсулина варьировали от 13 до 37 мМЕ/л (24,46 ± 9,2 мМЕ/л), у 55% детей отмечалась гиперинсулинемия (норма 3–17 мМЕ/л). Значения индексов И/Г (3,18 ± 1,22) и HOMAIR (6,45 ± 1,20) подтверждали состояние ИР у обследованных. Учитывая патогенетическую связь развития ИР с ожирением, мы оценили показатели инсулина в двух подгруппах больных: с нормальной массой тела и ожирением. У пациентов с ожирением гиперинсулинемия отмечалась в 44% случаев (4/9), с нормальной массой тела – в 62,5% (5/8). При нормальных значениях инсулина независимо от массы тела развитие заболевания связано с нарушением секреторной функции поджелудочной железы. Полученные результаты указывают на сочетанный характер ИР в генезе СД II у детей: связь физиологической (пубертатной) ИР и ИР, обусловленной дисфункцией β-клеток поджелудочной железы.
Протоколы лечения детей с СД II включают индивидуальную и семейную психологическую терапию, изменение образа жизни ребенка с достижением адекватной физической активности и рационального сбалансированного питания, обучение пациента и его семьи [16, 27, 29]. Приоритетной целью терапевтического воздействия является оптимальный гликемический контроль: уровень гликированного гемоглобина < 6,5%, значения тощаковой глюкозы < 126 мг/дл [25]. Однако достижение целевых показателей гликемии у детей представляет сложную задачу, связанную с физиологическими и психологическими возрастными особенностями. По мнению ряда авторов, не более 8–10% детей с СД II достигают оптимальных цифр гликемического контроля без дополнительной медикаментозной поддержки [25, 39, 43].
Сохранение нарушений углеводного обмена в детском возрасте увеличивает риск развития сердечно-сосудистых заболеваний и дисметаболических проявлений диабета [11, 12, 25]. Это определяет долгосрочность дополнительного использования в педиатрической практике фармакологических средств, корригирующих ИР. Для нормализации гликемии у детей при СД II рекомендуется использовать метформина гидрохлорид в виде моно- или комбинированной терапии с инсулином [5, 7, 13, 25, 29, 37, 46].
С 2007 г. для лечения детей с 10 лет разрешен к применению препарат «Сиофор 500/850» (метформина гидрохлорид, «Берлин-Хеми») [18]. Обычная стартовая доза в детском возрасте составляет 500 или 850 мг однократно. Коррекция дозы метформина проводится на основании показателей гликемии через 10–15 дней до достижения максимальной рекомендуемой суточной дозы препарата (2000 мг). Острые обратимые побочные эффекты метформина в отношении желудочно-кишечного тракта можно уменьшить до минимума путем приема лекарственного средства с едой или после еды, а также применением меньших дозировок, которые медленно увеличиваются [5].
Основной механизм действия метформина заключается в снижении продукции глюкозы печенью через активацию рецептора инсулина, как правило, посредством субстрата-2 инсулинового рецептора. В настоящее время влияние метформина на метаболизм глюкозы оценивается преимущественно как антигипергликемическое действие. Снижение показателей гликемии при использовании препарата не связано с повышением концентрации инсулина в плазме крови. Наблюдаемое снижение массы тела обусловлено анорексигенным эффектом. Отмечается уменьшение жирового депо (больше подкожного, чем висцерального) [25, 29].
Метформин улучшает периферическую чувствительность к инсулину. Снижение периферической ИР приводит к нормализации утилизации и метаболизма глюкозы в печени, мышцах и жировой ткани, что предотвращает развитие гипергликемии и клинических проявлений СД II у детей.
К настоящему времени завершено три рандомизированных двойных слепых плацебо-контролируемых исследования применения метформина у детей с ИР, нормальной толерантностью к глюкозе и наличием отягощенного семейного анамнеза по СД II [21, 26, 40]. В первой работе у 13 больных с ожирением и ИР установлено уменьшение по сравнению с плацебо массы тела (- 4,35 кг, Р = 0,02), ИМТ (- 1,26 кг/м2, Р = 0,002), окружности талии (- 2,8 см, Р = 0,003), подкожного жирового депо (-52,5 см2, Р = 0,002) и уровней базального инсулина (-2,2 мЕД/л, Р = 0,011). Чувствительность к инсулину улучшилась у 45% детей на фоне приема препарата и у 27% пациентов группы контроля (Р = 0,21) [40]. Результаты другого исследования (n= 29) показали снижение ИМТ (3,6% относительно группы контроля), показателей лептинемии, базальных уровней глюкозы (-0,54 ммоль/л) и инсулина (-83,3 пмоль/л) у больных даже при отсутствии ограничения пищевого калоража [21]. При изучении влияния метформина у детей с ожирением на фоне низкокалорийной диеты отмечено снижение массы тела на 2,7% относительно контроля, а также концентраций лептина, инсулина, глюкозы [26].
Проведенное многоцентровое контролируемое исследование эффективности и безопасности метформина у детей с СД II подтвердило улучшение гликемического контроля через 2 недели от начала терапии со снижением уровней базальной глюкозы на 42,9 мг/дл и гликированного гемоглобина на 1,4% от исходных значений по сравнению с группой сравнения (P < 0,001) [24].
Разработанный нами алгоритм лечения больных с СД II включает соблюдение диеты с суточным калоражем 1800–1900 ккал, ограничением жиров до 25% от общей калорийности и легкоусвояемых углеводов, ежедневную физическую нагрузку, применение сахароснижающих препаратов – бигуанидов (метформин гидрохлорида) и (или) генноинженерного инсулина (пролонгированного и (или) короткого действия) (рисунок).
У детей начальная доза метформина составляла 500 мг 1 раз в сутки после ужина. Назначение препарата проводилось в режиме титрации (по 500 мг) еженедельно. Максимальная доза достигала 2 000 мг в сутки (2–3 приема).
Через 3 месяца монотерапии метформином определяли уровень HbA1c. При сохранении показателей HbA1c > 7,0% (или ФА > 315 мкмоль/л) и гликемии натощак > 6,1 ммоль/л дополнительно назначали инсулин пролонгированного действия (инсулин НПХ (нейтральный протамин Хагедорна)) или аналоги инсулина пролонгированного действия. Повторно контролировали значения HbA1c через 3 месяца. При отсутствии достижения целевых уровней гликемического контроля (базальная гликемия: цельная капиллярная кровь – 3,5–6,1 ммоль/л или плазма – 3,89–7,22 ммоль/л, постпрандиальная гликемия до 8,9 ммоль/л, гликемия перед сном до 6,1 ммоль/л), повышенных показателях гликированных белков больного переводили на базис-болюсный режим инсулинотерапии (три инъекции инсулина короткого (аналог инсулина ультракороткого действия) действия перед основными приемами пищи и две инъекции инсулина НПХ или аналогов инсулина пролонгированного действия) с отменой метформина.
75% обследованных нами детей находились на монотерапии метформином (500–2000 мг/сут, средняя доза 1356,3 ± 643,8 мг/сут). Перевод на комбинированную терапию (метформин + инсулин (аналог) пролонгированного действия) осуществлен у 20% пациентов ввиду недостаточного контроля гликемии при лечении метформином. Один больной СД II c синдромом Прадера–Вилли находился на базис-болюсной инсулинотерапии. Установленное нами соотношение схем лечения СД II типа отличается от данных J.H. Silverstein et al., отметивших использование у 44% детей пероральных сахароснижающих препаратов и у 48% – инсулина [38]. Выявленное различие может быть обусловлено относительно малым сроком наблюдения за обследованными больными.
Оценку степени метаболической компенсации проводили ежемесячно на протяжении 12 месяцев наблюдения: показатели ФА колебались от 204 до 435 мкмоль/л на фоне коррекции схем терапии сахароснижающими препаратами. У детей, получающих только метформин, средние уровни ФА составляли 308,9 ± 15,8 мкмоль/л. При комбинированном лечении показатели гликированного белка соответствовали 253,9 ± 13,6 мкмоль/л, при инсулинотерапии – 269,5 ± 11,0 мкмоль/л и были достоверно ниже значений ФА при монотерапии пероральными препаратами (P < 0,05).
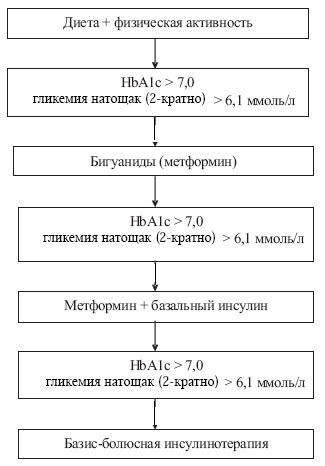
Рис. Алгоритм лечения CД II у детей
Таким образом, на момент верификации диагноза СД II у обследованных детей выявлен феномен инсулинорезистентности, отражаемый достоверным повышением индексов И/Г и HOMAIR. Степень выраженности описанных нарушений углеводного обмена варьировала индивидуально и была обусловлена сочетанием физиологической (пубертатной) ИР и ИР, обусловленной дисфункцией β-клеток поджелудочной железы. На фоне оптимизации индивидуального лечебного подхода с применением разных схем терапии отмечено достижение оптимального гликемического контроля по уровням гликированного белка у всех пациентов. Необходимо отметить, что долгосрочным результатом эффективности комплексного лечения СД II у детей должно стать снижение частоты случаев развития и прогрессирования поздних осложнений заболевания, ведущих к ранней инвалидизации и смертности больных.
1. Князев Ю.А. Возрастные гормонально-метаболические нормативы: науч.-метод. пособие для педиатров и эндокринологов. – М., 1998.
2. Кураева Т.Л. // Сахарный диабет. – 2005. – № 3. – С. 14 –16.
3. Малявская С.И., Дворяшина И.В., Терновская В.А. Метаболический инсулинорезистентный синдром: диагностика, клиническое значение, педиатрические аспекты. – Архангельск: Сев. гос. мед. университет, 2004.
4. β-клетка: секреция инсулина в норме и патологии / под ред. И.И. Дедова. – М., 2006.
5. Сворен Б.М., Вульфсдорф Д.И. // Intern. Diabetes Monitor. – 2006. – Vol. 18, N 5, 6. – P. 1 – 14.
6. ADA // Pediatrics. – 2000. – Vol. 105. – P. 671–680.
7. ADA consensus statement. Type 2 diabetes in children and adolescents // Diab. Care. – 2002. – Vol. 25. – P. 89–94.
8. Arslanian S.A., KalhanS.C. // Diabetes. – 1994. – Vol. 43. – P. 908–914.
9. Aye T., Levitsky L.L. // Curr. Opin. Pediatr. – 2003. – Vol. 15. – P. 411–415.
10. Banerji M.A., ChikenR.I., Huey H. et al. // Diabetes. – 1994. – Vol. 43. – P. 741–745.
11. Caprio S., Tamborlane W.V. // Endocrinol. Metab. Clin. North Amer. – 1999. – Vol. 28. – P. 731–747.
12. Caprio S. // J. Pediatr. Endocrinol. Metab. – 2002. – Vol. 15. – P. 487–492.
13. Castells S. // J. Pediatr. Endocrinol. Metab. – 2002. – Vol. 15 (suppl. 1). – P. 531–540.
14. Cho N.H., Silverman B.L., Rizzo T.A. // J. Pediatr. – 2000. – Vol. 136. – P. 587–592.
15. Cole T.J., Bellizzi M.C., Flegal K.M. et al. // BMJ. – 2000. – Vol. 320. – P. 1240–1243.
16. Daniels S.R. // Curr. Atheroscler. Rep. – 2001. – Vol. 3. – P. 479–485.
17. Deckelbaum R.J., Williams C.L. // Obes. Rev. – 2001. – Vol. 9. – P. 239–243.
18. Eis E.B. // Deutsches Artzteblatt. – 2007. – H.1–2. – S. A64.
19. Eriksson J.G., Forsen T., Tuomilehto J. et al. // BMJ. – 1999. – Vol. 318. – P. 427–431.
20. Eriksson J.G., Forsen T., Tuomilehto J. et al. // BMJ. – 2001. – Vol. 322. – P. 949–953.
21. Freemark M., Bursey D. // Pediatrics. – 2001. – Vol. 107. – E55.
22. Haffner S.M., Miettinen H., Gaskell S.P. et al. // Diabetologia. – 1996. – Vol. 39. – P. 1201–1207.
23. Jaquet D., Gaboriaau A., Czernichow P. et al. // J. Clin. Endocrinol. Metab. – 2000. – Vol. 85. – P. 1401–1406.
24. Jones K.L., Arslanian S., Peterkova V.A. et al. // Diabetes Care. – 2002. – Vol. 25. – P. 89–94.
25. Kaufmann F. R. // Endocrin. Metab. Disorders. – 2003. – Vol. 4. – P. 33–42.
26. Kay J.P., Alemzadeh R., Langley G. et al. // Metabolism. – 2001. – Vol. 50. – P.1457–1461.
27. Kiess W., Galler A., Reich A. // Obes. Rev. – 2001. – Vol. 2. – P. 29–36.
28. Kiess W., Boettner A. // Adolesc. Med. – 2002. – Vol. 13. – P. 181–190.
29. Kiess W., Boettner A., Raile K. et al. // Horm. Res. – 2003. – Vol. 59 (suppl. 1). – P. 77–84.
30. Levy-Marchal C., Jaquet D. // Ped. Diabetes. – 2004. – Vol. 5. – P. 147–153.
31. Lucas A., Fewtrell M.S., Cole T.J. // BMJ. – 1999. – Vol. 319. – P. 245–249.
32. Morrison J.A., Sprecher D.l., Barton B.A. et al. // J. Pediatr. – 1999. – Vol. 135.
33. Neel J.V. // Amer. J. Hum. Genet. – 1962. – Vol. 14. – P. 353–362.
34. Ong K.K., Ahmed M.L., Emmet P.M. et al. // BMJ. – 2001. – Vol. 322. – P. 949–953.
35. Quon M.J. // J. Clin. Endocrinol. Metab. – 2002. – Vol. 87. – P. 650–654.
36. Ranke M. B. Diagnostics of Endocrine Function in Children and Adolescents. – Leipzig: Heidelberg, 1996.
37. Rosenbloom A.L. // Pediatric Drugs. – 2002. – Vol. 4. – P. 209–221.
38. Silverstein J.H., Rosenbloom A.L. // J. Pediatr.Endocrinol. Metab. – 2000. – Vol. 13 (suppl. 1). – P. 1–7.
39. Silverstein J.H., Rosenbloom A.L. // Curr. Diab. Report. – 2001. – Vol. 1. – P. 19–27.
40. Srinivasan S., Ambler G.R., Baur L.A. et al. // J. Clin. Endocrinol. Metab. – 2006. – Vol. 91. – P. 2074–2080.
41. Stolecke H. Nosologische, metabolische und endokrinologische Aspekte der Adipositas. – Berlin; Heidelberg, 1997.
42. Stumvoll M., Jacob S. // Exp. Clin. Endocrinol. Diabetes. – 1999. – Vol. 107. – P. 1643–1648.
43. Viberti G., Kahn S.E., Greene D.A. et al. // Diab. Care. – 2002. – Vol. 25. – P. 1737–1743.
44. Wabitsch M. // Eur. J. Pediatr. – 2000. – Vol. 159. – P. 8–13.
45. Wallace T.M., Levy J.C., Matthews D.R. // Diab. Care. – 2004. – Vol. 27. – P. 1487–1495.
46. Zuhri-Yafi M.I., Brosnan P.G., Hardin D.S. // J. Pediatr. Endocrinol. Metab. – 2002. – Vol. 15 (suppl. 1). – P. 541–546.
Медицинские новости. – 2008. – №14. – С. 87-91.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.