Внимание! Статья адресована врачам-специалистам
Mokhort T.V.
Belarusian State Medical University, Minsk
Cognitive function and diabetes mellitus:
what should know the clinician?
Резюме. До настоящего времени проблема оценки состояния когнитивной функции при сахарном диабете не являлась приоритетной. Однако многочисленные исследования позволили оценить влияние различных патогенетических факторов на развитие когнитивного дефицита. Рассмотрено влияние на прогрессию снижения когнитивной функции гипергликемии, сосудистых и клеточных нарушений, ожирения, ассоциированного с инсулинорезистентностью, дефицитов нейротрофических мозговых факторов, маркеров воспаления, церамидов, лептинрезистентностью. Проведен анализ патогенетической связи между сахарным диабетом 2-го типа и болезнью Альцгеймера. Обосновано негативное влияние на когнитивный дефицит гипогликемических эпизодов. Подробно проанализированы терапевтические возможности предупреждения и лечения нарушений когнитивной функции при сахарном диабете.
Ключевые слова: сахарный диабет, когнитивный дефицит, болезнь Альцгеймера, гипогликемии.
Медицинские новости. – 2018. – №10. – С. 13–20.
Summary. Up to the present time, the problem of assessing cognitive function in diabetes mellitus has not been a priority. However, many studies have made it possible to evaluate the influence of different pathogenetic factors on the development of cognitive deficits. Influences on the progression of cognitive decline in hyperglycemia, vascular and cellular alterations, obesity associated with insulinresistance, deficits of neurotrophic brain factors, markers of inflammation, ceramides, leptinresistance are considered. An analysis of the pathogenetic connection between type 2 diabetes mellitus and Alzheimer’s disease was carried out. The negative impact on the cognitive deficiency of hypoglycemic episodes is substantiated. The detail analyze of therapeutic possibilities of preventing and treating cognitive impairment in diabetes mellitus is given.
Keywords: diabetes mellitus, cognitive deficiency, Alzheimer’s disease, hypoglycemia.
Meditsinskie novosti. – 2018. – N10. – P. 13–20.
Сахарный диабет (СД) прочно ассоциируется у врачей различных специальностей с увеличением риска развития:
· сердечно-сосудистой патологии и смертности от нее;
· слепоты вследствие диабетической ретинопатии;
· прогрессивного снижения функции почек, обусловленного диабетической нефропатией;
· ампутаций в результате развития диабетической невропатии или облитерирующих поражений сосудов конечностей.
В то же время на протяжении многих лет, несмотря на общность механизмов развития ангио- и невропатий, не уделялось достаточного внимания определению состояния головного мозга с позиций оценки когнитивной функции. До настоящего времени не разработано обоснованных критериев диагностики нарушений функции мозга при СД, несмотря на исторические факты. В 1922 году W.R. Miles и H.F. Root доказали связь между СД и когнитивной дисфункцией, а в 1963 году E. Reske-Nielsen и соавт. при посмертном исследовании 16 молодых пациентов, длительно болеющих СД 1-го типа, выявили нейропатологические изменения мозга – фиброз оболочек, ангиопатию, псевдокальцинаты и выраженную диффузную дегенерацию белого и серого вещества – и сделали заключение, что эти патологические изменения являются результатом СД и могут быть обозначены как диабетическая энцефалопатия [46]. Позже была установлена связь СД с увеличением распространенности случаев инфаркта мозга во всех возрастных группах и установлена обратная корреляция между когнитивными функциями и плотностью белого вещества мозга, атрофией мозга и наличием очагов в веществе мозга, строго ассоциированная с уровнем HbA1 и длительностью СД по результатам магнитно-резонансной томографии [38].
Исторически проблемы когнитивных нарушений рассматривались с позиций оценки понятий «деменция» или «слабоумие», характеризующих наиболее тяжелые когнитивные нарушения, приводящие к дезадаптации в повседневной жизни. В настоящее время анализируются менее выраженные расстройства (легкие – при которых показатели психометрических шкал могут оставаться в пределах среднестатистической возрастной нормы или незначительно отклоняться при осознании пациентом снижения когнитивных способностей; умеренные – выходящие за рамки возрастной нормы, но не ограничивающие самостоятельности и независимости пациента).
Анализ связи деменции и СД свидетельствует о том, что риск развития деменции в среднем в 1,6–2,5 раза выше, сосудистой деменции – в 2,0–2,6 раза выше, болезни Альцгеймера – в 1,5 раза выше, чем в общей популяции, независимо от возраста начала СД [10].
Обоснование вовлечения в патологический процесс заключается в понимании того, что глюкоза – основной энергетический субстрат мозга, с другой стороны существуют неопровержимые факты, свидетельствующие о роли инсулина в регуляции функции мозга, а именно:
· рецепторы инсулина обнаружены на мембранах нейронов гипоталамической и лимбической зоны, гиппокампа;
· может поступать в структуры головного мозга путем активного транспорта через гемато-энцефалический барьер и синтезироваться локально в мозге;
· регулирует метаболизм глюкозы в нейронах мозга, обеспечивая его энергией;
· необходим в процессе консолидации информации, так как в гиппокампе глюкоза проникает в клетку инсулинозависимо, что повышает энергетический потенциал нейронов;
· является медиатором нейротрофических эффектов, усиливает обмен моноаминов, снижает холинергическую активность в нейронах стриатума;
· противодействует апоптозу нейронов [25, 26,39].
Развитие микрососудистых осложнений сопровождается нарушением структуры и функции мозга и, как следствие, приводит к нарушению когнитивной функции. Генез нарушения когнитивной функции при СД включает сочетанное влияние макро- и микроангиопатии и невропатии, акселерацию атеросклероза и старения, развития болезни Альцгеймера.
На рисунке 1 представлена общая схема развития микрососудистых осложнений, определяющая системное влияние гипергликемии на микроциркуляторное русло и нейроны [8], но для каждой локализации анализируются различные особенности и дополнительные факторы, определяющие нарушение функции органа.
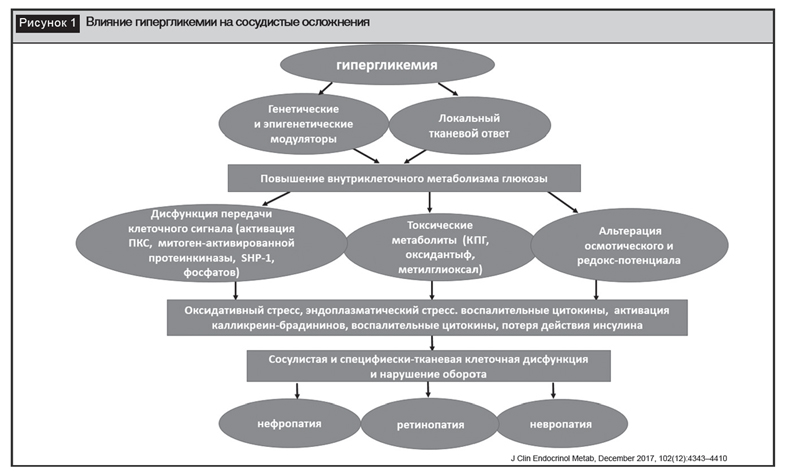
Механизм развития нарушений функции мозга включает влияние множества факторов.
Гипергликемия
Влияние гипергликемии реализуется через нарушение передачи клеточного сигнала за счет активации протеинкиназы С, митоген-активированной протеинкиназы, протеиновой тирозин фосфатазы SHP-1, фосфатов, накопления токсических метаболитов (конечных продуктов гликирования (КПГ), метилглиоксала, прооксидантов) и нарушений осмотического состояния клеток и редокс-потенциала, которые активируют оксидативный и эндоплазматический стресс, провоспалительных цитокинов, калликреин-брадикининовой системы и снижения действия инсулина. Доказано, что КПГ накапливаются в нейрофибриллярных клубках и амилоидных бляшках в мозге, способствуя развитию болезни Альцгеймера (БА) [13]. В то же время активация оксидативного стресса через формирование активных форм кислорода приводит к митохондриальной дисфункции и структурным изменениям в b-амилоиде [16]. Гипотеза cвязи БА и СД базируется на научных фактах и наличии общих черт, таких как рост заболеваемости с возрастом, доказательства генетической предрасположенности и сопоставимые патологические особенности в b-островках и головном мозге (депозиты b-амилоида в головном мозге при БА и в островках поджелудочной железы при СД 2-го типа), накопление КПГ в нейрофибриллярных клубках и амилоидных бляшках в мозге. Общность этих патологических процессов привела к предположению о наличии СД 3-го типа, характеризующегося:
– патологией функции инсулина на уровне мозга (нарушение высвобождения нейротрансмиттеров в синапсах и нарушение сигнальных путей, связанных с обучением и долгосрочной памятью);
– выявлением обратно пропорциональной зависимости между экспрессией инсулина и выраженности развития БА;
– развитием инсулинорезистентности (ИР) вследствие увеличения продукции нейротоксинов, продуцируемых амилоидом (amyloid beta-derived diffusible ligands – ADDLs), которые уменьшают пластичность синапсов, способствуют окислительному повреждению и гиперфосфорилированию Tau-протеинов и нарушают трансдукцию сигнала в синапсах;
– снижением клиренса??-амилоида [41].
До настоящего времени нет однозначного ответа на вопрос, что первично: нарушения гомеостаза глюкозы или формирование амиолоидных бляшек. Существуют доказательства связи распространенности БА и СД. Так, по результатам различных исследований определено, что наличие СД приводит к удвоению риска развития БА. Доказано, что декомпенсация СД повышает риск БА. Механизм влияния декомпенсации СД связывают с нейротоксическим влиянием КПГ, инициирующим гиперфосфорилирование Tau-протеина и аккумуляцию ?-амилоида (рис. 2) [58]. В то жевремя у пациентов с БАсущественночащевыявляютсянарушенияуглеводногообмена (предиабета иСД) [30]. Однако связь когнитивных нарушений и БА с СД не вызывает сомнений.
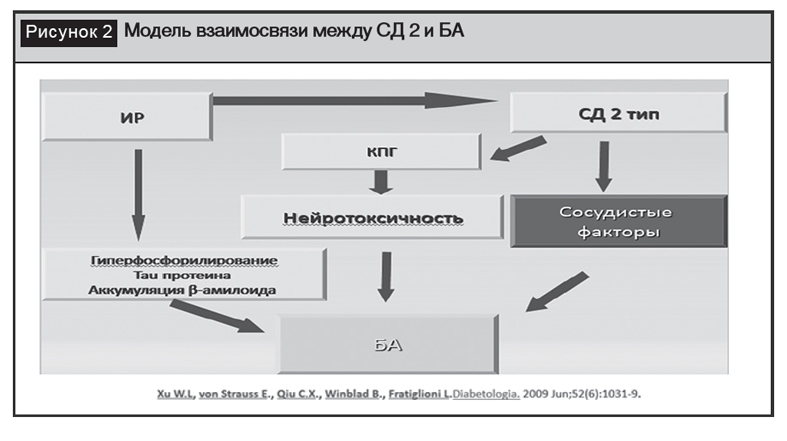
Изменение структуры мозга вследствие сосудистых и клеточных нарушений
К настоящему времени, бесспорно, доказано наличие структурных нарушений в мозге, которые выявляются при использовании современных визуализационных технологий.
Выраженность сосудистых нарушений характеризуется изменениями белого вещества головного мозга, а также развитием лейкоареоза – снижением плотности белого вещества головного мозга на КТ или повышением при МРТ в парагиппокампе, височной и лобной областях. Начальные изменения развиваются вокруг передних рогов боковых желудочков, распространяясь далее по боковым отделам желудочков симметрично в обоих полушариях мозга, и нарастают с увеличением возраста. Параллельно регистрируются уменьшение объема мозга, единичные или множественные немые инфаркты мозга, повышение общего объема цереброспинальной жидкости с потенциальным формированием гидроцефалии, снижение плотности серого вещества в лобных и теменных долях, корковая атрофия [42, 54]. Мета-анализ 50 исследований по визуализации мозга подтвердил большую выраженность сосудистых нарушений, гиперинтенсивности белого вещества, повышение частоты лакунарных инфарк-тов и церебральной атрофии [54].
Влияние образа жизни на дополнительные факторы, определяющие нарушение функции и структуры мозга
В течение многих лет отмечалась связь между когнитивными нарушениями и массой тела (рис. 3) [56]. Также доказано снижение объема гиппокампа при увеличении окружности талии и определено негативное влияние именно висцерального ожирения на развитие когнитивных нарушений. Модель, определяющая влияние висцерального ожирения на функции мозга, включает активацию факторов воспаления, снижение тромболитических свойств крови, развитие ИР, а также артериальную гипертензию и дислипидемию. Перечисленные механизмы реализуются через угнетение функции нейротрофического фактора мозга (brain-derived neurotrophic factor (BDNF) – белок человека, кодируемый геном) [50, 57].
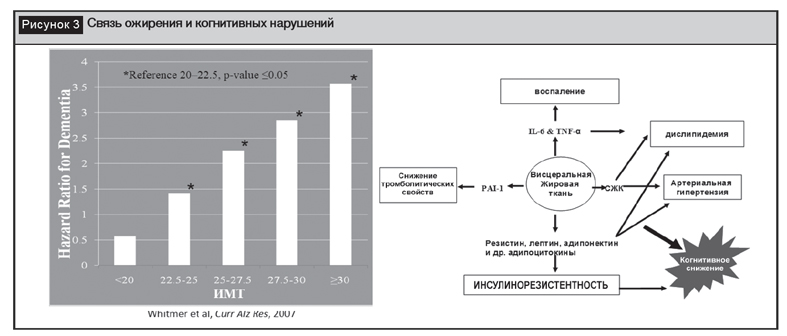
Когнитивная дисфункция при СД 2-го типа коррелирует с уровнем маркеров воспаления, которые могут также способствовать развитию БА, что подтверждено при проведении многих исследований [27, 47]. В качестве альтернативного механизма обсуждается механизм инициации когнитивных нарушений через патологическиеизменения в гипоталамо-гипофизарно-надпочечниковой оси и повышение уровня кортизола [22].
Одним из механизмов, определяющих снижение когнитивной функции при СД 2-го типа и связывающих вышеперечисленные нарушения, является увеличение синтеза церамидов, которое ассоциировано с резистентностью к лептину и инсулину. Повышенный уровень церамидов приводит к ингибированию пути трансдукции сигнала инсулина и фосфорилированию серина JNK (c-Jun N-terminal kinases), что усугубляет ИР. Церамиды являются токсичными липидами, которые, синтезируясь в печени, могут проникать через гемато-энцефалический барьер и оказывать влияние на метаболические процессы в мозге, нарушая процессы фосфорилирования и активируя провоспалительные цитокины. Исследования с мечеными церамидами продемонстрировали нейрональную резистентность к инсулину, активацию оксидативного стресса, усугубление митохондриальной дисфункции, нарушение холинергической передачи, повреждение ДНК, активацию перекисного окисления липидов и экспрессию белка-предшественника амилоида (APP) и др. нарушения (активация пути PI3K-Akt и стимулированного IGF -1 энергетического метаболизма), лежащие в основе нейродегенерации и формирования когнитивного дефицита [7, 52]. Реализация этих нарушений проходит через нарушение пластичности олигоденроцитов и образования миелина в головном мозге и активацию локальной продукции церамида в мозге, замыкая порочный круг [52].
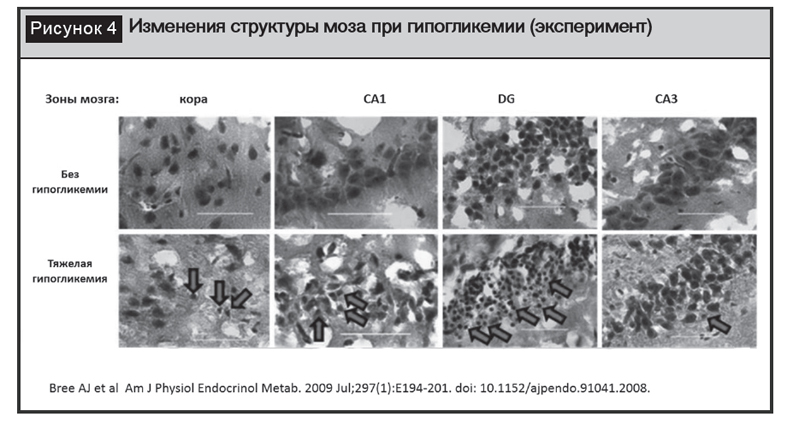
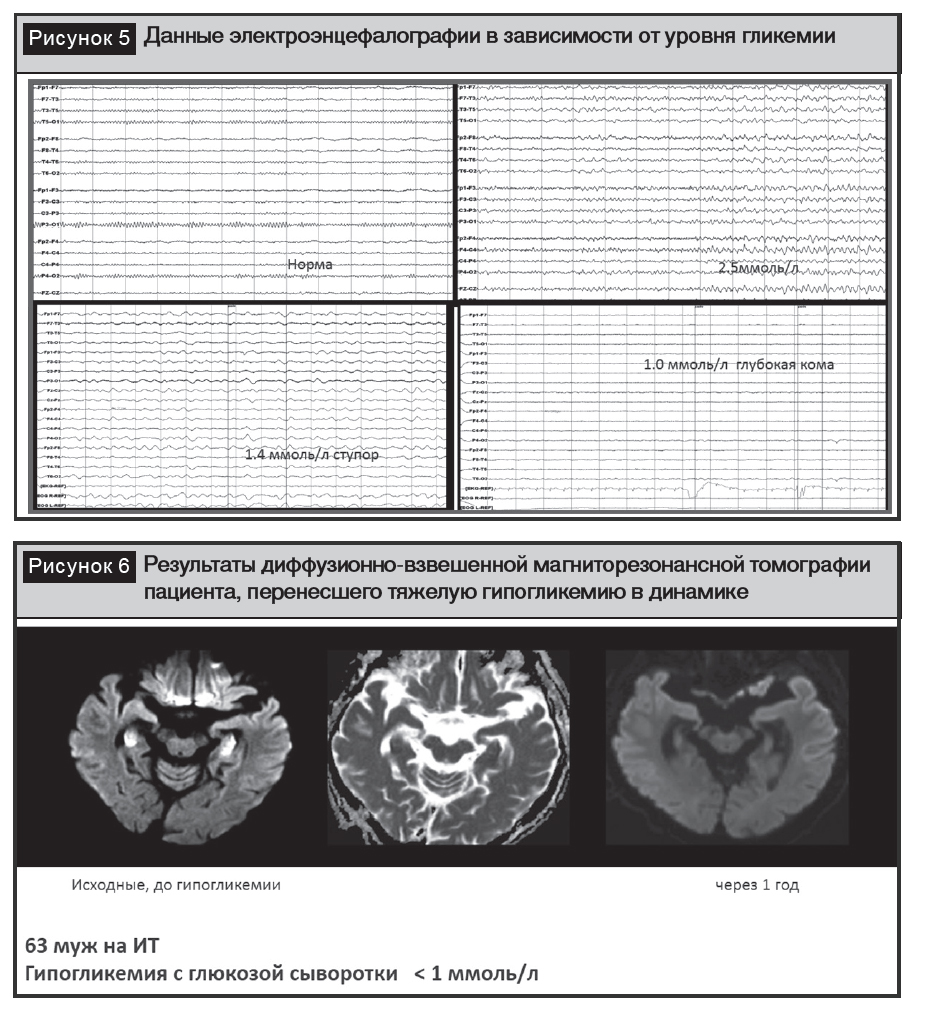
Продолжение развития патологических изменений в мозге связано с тем, что инсулин регулирует потоки нейротрофических факторов, что не обеспечивает адекватные процессы нейрорегенерации, с другой стороны, ИР, являющаяся маркером СД 2-го типа, сопровождается снижением церебральной перфузии и при развитии гипогликемии – нарушением потребления глюкозы клетками мозга. Экспериментальные исследования продемонстрировали выраженные нарушения структуры мозга у животных при развитии гипогликемии (рис. 4) [42]. В клинической практике гипогликемии проявляются нейроглюкопении, что характеризуется изменениями на электроэнцефалограмме, используемой в качестве дополнительного диагностического теста (рис. 5). При использовании диффузионно-взвешенной магнитно-резонансной томографии, основанной на регистрации скорости перемещения отмеченных радиоимпульсами протонов, позволяющей характеризовать сохранность мембран клеток и состояние межклеточных пространств, также выявляются выраженные изменения при динамическом наблюдении после глубокой гипогликемии (рис. 6). Долгое время спорным считался вопрос о влиянии легкой гипогликемии при СД 2-го типа на когнитивную функцию, но к настоящему моменту при наблюдении за 15 404 пациентами (средний возраст – 62,4 года) у 7,2% развилась деменция, причем заболеваемость деменцией оказалась достоверно выше у пациентов с зарегистрированными гипогликемическими эпизодами. Дополнительным фактором, усугубляющим развитие когнитивных расстройств, могут быть дисметаболические патологические состояния, включающие гипотиреоз, нарушение функции печени или почек, дефицит витамина В12, фолиевой кислоты и др., сопутствующие СД. С этих позиций особое внимание вызывает дефицит витамина В12, развивающийся на фоне длительного использования метформина [6]. Параллельно доказана связь дефицита витамина В12, повышения уровня гомоцистеина со снижением когнитивной функции мозга и уменьшением общего объема головного мозга [18].
Когнитивные функции – наиболее сложные функции мозга, обеспечивающие процесс познавания мира и взаимодействия с ним, восприятие информации. Они включают обработку и анализ, запоминание и хранение информации, обеспечивают ее обмен, построение и осуществление программы действий.Для оценки когнитивных функций используются нейропсихологические методы исследования, которые представляют собой различные тесты и пробы на запоминание и воспроизведение слов и рисунков, узнавание образов, решение интеллектуальных задач, исследование движений и т. д. (тест рисования часов, тест 5 слов, краткая шкала MMSE (Mini-mental State Examination), шкала – Батарея лобной дисфункции (Frontal Assessment Batter), Монреальская шкала когнитивной оценки (МоСА), Digit Symbol Substitution Test DSST (ранее стандартизованные тесты DANTES), тест Струпа (Stroop effect), ReyAuditoryVerbalLearningTest (RAVLT) и др.) и позволяют оценить степень когнитивного дефицита.
В клинических наблюдениях по результатам различных тестов определено, что повторные эпизоды тяжелой гипогликемии, потребовавшие проведение неотложной медицинской помощи, повышают риск развития деменции в 2,4 раза у пациентов с СД 2-го типа [34, 44]. В настоящее время при СД 1-го типа связь гипогликемии с когнитивным дефицитом не вызывает сомнений, но при СД 2-го типа, особенно в условиях лечения таблетированными препаратами, вопрос связи гипогликемии, особенно легкой, не требующей медицинский помощи, с развитием деменции остается спорным в связи с тем, что на когнитивную функцию оказывают влияние множество факторов, кроме уровня гликемии (наследственность, возраст (точнее, старение), курение, тип питания, физическая активность, режим дня и поведенческие особенности, артериальная гипертензия, дислипидемия, наличие микроангиопатий и др.).
При патогенетической обоснованности и доказанности когнитивных нарушений при СД, обусловленных не только функциональными, но и структурными изменениями мозга, правомочно возникает вопрос: какие опции могут быть использованы для минимизации когнитивного дефицита и может ли контроль за течением СД их предупредить? В настоящее время определены с различной степенью доказательности потенциальные действия по обеспечению сохранности когнитивной функции, включающие:
· компенсацию углеводного обмена с поддержанием максимальной нормогликемии без гипогликемий;
· нормализацию массы тела;
· депривацию курения и алкоголя;
· умственные тренировки – так называемый фитнесс для мозга;
· музыкотерапию;
· физическую активность;
· нормализацию артериального давления и профиля липидов;
· использование антиоксидантов и нейропротекторов.
Выбор антигипергликемической терапии, как указывалось выше, должен быть основан на обеспечении целевых уровней гликемии без гипогликемических эпизодов. К настоящему времени нет доказательной базы, основанной на результатах рандомизированных клинических исследований, позволяющей определить оптимальные антигипергликемические средства, однако имеются публикации, определяющие преимущества некоторых классов препаратов. Так, препарат первого выбора в лечении СД 2-го типа – метформин. Важно знать, что он обеспечивает уменьшение проявлений нейропатологии мозга, подобных изменениям при БА у лептин-резистентных мышей, выбранных в качестве модели СД 2-го типа [33].
Второй класс препаратов, имеющий потенциальные бенефиции, – тиазолидиндионы, точнее, единственный представитель, использование которого разрешено в настоящее время, а именно пиоглитазон, который уменьшает дефицит синаптической передачи и обеспечивает долговременное потенцирование, уменьшая церебральную дисфункцию в эксперименте [14, 51, 59]. Аналогичные результаты получены в плацебо-контролируемом слепом клиническом исследовании, проведенном в параллельных группах, в котором продемонстрировано улучшение когнитивной функции при использовании розиглитазона в течение 6 месяцев.
Представляют интерес доказательства потенциала инкретин-ассоциированной терапии. Доказано, что инкретины увеличивают синаптическую пластичность, клеточную пролиферацию, память, уменьшают отложение A?42-бляшек, выраженность оксидативного стресса, активность воспаления и проявления глиоза в различных экспериментальных моделях, обеспечивая нейропротективный и нейрорегенераторный эффекты [23, 31, 43].
Роль инсулина в обеспечении энергетических потребностей мозга известна и бесспорна, однако инсулинотерапия ассоциирована с увеличением вероятности развития гипогликемических эпизодов, риск которых растет при развитии когнитивных нарушений, ограничивающих самоконтроль. Тем не менее доказано, что инсулин и инсулиновые факторы роста, в частноcти ИФР1, поддерживают выживание олигодендроглиальных и нейрональных клеток. Альтернативным и оптимальным путем поступления инсулина является ингаляционный, так как ингаляционный инсулин способен лучше проникать через гемато-энцефалический барьер [48]. Данная форма инсулина доступна в мире, но опыт ее использования ограничен и лимитирован высокой стоимостью препарата, противопоказаниями и побочными эффектами.
Роль сосудистого фактора и артериальной гипертензии в развитии нарушений функции мозга определяет потенциальные бенефиции от нормализации артериального давления. В разделе исследования HYVET, посвященном когнитивным функциям у больных артериальной гипертензией (HYVETCOG), было доказано улучшение когнитивных функций при нормализации артериального давления [45], а каждый год антигипертензивной терапии с поддержанием целевых уровней артериального давления снижает риск заболеваемости деменцией различной этиологии. При этом определено, что снижение систолического АД <130 мм рт. ст. ассоциировано доказанным повышением риска развития ишемических инсультов, что определяет целевые уровни артериального давления – менее 140/90 мм рт. ст.
Фактором, ассоциированным с когнитивными нарушениями, выступает дислипидемия. Препаратами первого выбора в лечении дислипидемии являются статины, использование которых связывается с потенциально негативным влиянием на когнитивные функции, что определяет требования регулирующих органов FDA (США) и MHRA (Великобритания) о внесении в инструкции к этим препаратам предостережений о возможных негативных последствиях. Данные Кохрановского мета-анализа не позволяют сделать заключение о протективном влиянии статинов на развитие когнитивного снижения различной этиологии, но не свидетельствуют и об ухудшении когнитивных нарушений [40].Одним из крупных многоцентровых рандомизированных исследований с субисследованием Heart Outcomes Prevention Evaluation-3 (HOPE -3), включающем наблюдение за более чем 12000 пациентов в возрасте 70 лет и старше с промежуточным уровнем риска сердечно-сосудистой патологии, доказано отсутствие влияния на функции мозга в течение 5–6 лет при использовании розувастатина по динамике чувствительной оценочной шкалы нейропсихологической функции (Digit Symbol Substitution Test – DSST) [11, 35]. Отдельно следует отметить, что агонисты рецептора пролифератора пероксисом?? (PPAR-?) – фибраты – в экспериментальных исследованиях на животных обеспечивают нейропротективные эффекты, проявляющиеся в прямом влиянии на нейрогенез в области гиппокампа и замедлении активации микроглии, что в итоге приводит к улучшению координации и когнитивных функций у животных [9].
Также обсуждается польза включения в рацион жирной рыбы, содержащей большое количество ?-3-полиненасыщенных жирных кислот, не менее 5 раз в неделю на сохранение эффективной деятельности мозга [14].
Наконец, для оптимизации функции мозга потенциально могут быть использованы нейроцитопротекторы – лекарственные средства, обеспечивающие защиту нервных клеток от патогенных факторов, которые устраняют или уменьшают патофизиологические и биохимические нарушения в нервных клетках, уменьшают развитие тяжелых и необратимых повреждений нейронов при негативных воздействиях на нейроны. Из многообразия препаратов этого класса, включающего ноотропы, антиоксиданты, препараты, улучшающие кровообращение мозга, препараты комбинированного действия, адаптогены и др., следует отметить, что, к сожалению, использование нейроцитопротекторов для коррекции когнитивных нарушений при СД ограничено.
На стадии умеренных и легких когнитивных нарушений главной целью терапии является не столько улучшение памяти, сколько предотвращение прогрессирования когнитивных расстройств, поэтому препаратами первого выбора являются лекарственные средства с нейропротективным эффектом, который предполагается у так называемых сосудистых и метаболических препаратов.
Препараты, оказывающие преимущественно влияние на сосуды и мозговой кровоток, включают:
· ингибиторы фосфодиэстеразы: эуфиллин, пентоксифиллин, винпоцетин, препараты гинкго билоба и др.;
· блокаторы кальциевых каналов: циннаризин, флюнаризин, нимодипин, оказывающие вазодилатирующий эффект благодаря уменьшению внутриклеточного содержания кальция в гладкомышечных клетках сосудистой стенки, особенно в сосудах вертебрально-базилярного бассейна;
· блокаторы ?2-адренорецепторов: ницерголин (устраняет сосудосуживающее действие медиаторов симпатической нервной системы: адреналина и норадреналина).
Оптимизирующее действие на нейрометаболические процессы оказывают ГАМК-ергические препараты (пирацетам и его производные), пептидергические препараты и аминокислоты (церебролизин, актовегин, солкосерил, глицин, семакс). Эти препараты оказывают ноотропное действие, увеличивая пластичность нейронов головного мозга, и способствуют выживаемости нейронов в условиях гипоксии или при моделировании нейродегенеративного процесса.
Полифакторность изменений, сопровождающих когнитивные нарушения при СД, определяет потенциальное использование средств, которые способны воздействовать на различные аспекты патогенеза этой патологии. Альтернативным препаратом является хорошо известный и часто используемый в течение более 40 лет депротеинизированный гемодиализат из сыворотки телят Актовегин, который обладает мощным антиоксидантным и антигипоксантным действием и оказывает положительное влияние на когнитивные функции при СД и постинсультных когнитивных нарушениях, умеренных когнитивных нарушениях, улучшает психическую активность, интеллектуальную гибкость, устойчивость запоминания, оптимизирует регуляцию произвольной деятельности, что в целом обеспечивает улучшение общей активности и социальной адаптации пациентов [2, 3, 20]. Нейропротективные свойства Актовегина (поддержание жизнеспособности нейронов, уменьшение апоптоза, увеличение общего числа синаптических связей и снижение выраженности окислительного стресса) [21] обосновывают его применение при когнитивном дефиците.
Эффективность Актовегина является результатом его антигипоксантного действия, которое заключается в улучшении инсулин-независимого транспорта глюкозы в клетку путем активации действия инозитол фосфо-олигосахаридов, содержащихся в препарате, минуя инсулиновые рецепторы. Максимальный эффект по динамике показателей нейропсихологических тестов отмечен у пациентов с микроангиопатиями, то есть в условиях доказанной ишемии и активизации оксидативного стресса, так как Актовегин оптимизирует микроциркуляцию, способствует нормализации эндотелий-зависимых реакций и снижению периферического сосудистого сопротивления, минимизирует проявления оксидативного стресса, мобилизует поступление кислорода в клетки головного мозга и церебральных сосудов, улучшает реологические свойства крови [21, 37].
В настоящее время завершено крупное рандомизированное двойное слепое плацебо-контролируемое 12-месячное исследование терапевтической эффективности Актовегина – ARTEMIDA (A Randomized Trial of Efficacy, 12 Months International Double-blind Actovegin) – у 503 пациентов в возрасте старше 60 лет с постинсультными когнитивными нарушениями. Актовегин назначался peros по 2000 мг в сутки сроком до 20 дней и затем 1200 мг в сутки в течение 6 месяцев. Для оценки эффективности препарата проводилась оценка изменений когнитивного функционирования по шкалам ADAS-Cog+ и MoCA от исходных показателей через 3, 6 и 12 месяцев. Через 3 месяца по шкале MoCA и через 6 месяцев по шкале ADAS-Cog+ от начала лечения было доказано улучшение когнитивной функции, которое сохранялась после отмены лечения в течение последующих 6 месяцев наблюдения по обеим шкалам. Была также выявлена тенденция к уменьшению числа пациентов с диагнозом деменция в группе принимавших Актовегин по сравнению с группой, принимавших плацебо, в которой клинически верифицированная деменция выявлялась на 30% чаще [29]. Клиническая эффективность Актовегина обусловлена комплексным мультимодальным механизмом действия, включающим в себя антигипоксантный, нейропротективный эффекты и положительное воздействие на микроциркуляцию, что особенно актуально при СД.
Вариантом выбора коррекции снижения когнитивного дефицита является цитиколин – нуклеотид, незаменимый предшественник фосфатидилхолина (лецитина) – основного структурного компонента клеточных мембран, включая нейрональные, что определяет потенциал его использования в качестве нейропротектора, применяющегося при лечении заболеваний, сопровождающихся повреждением нейронов ишемического, травматического или дегенеративного характера. Цитиколин (Цераксон) способен актировать биосинтез фосфатидилхолина, поддерживать нормальный уровень кардиолипина и сфингомиелина, нормализовать холинергическую передачу и работу других нейротрансмиттерных систем в мозге, снижать концентрацию нейротоксичного глутамата, противодействовать глутамат-индуцированному апоптозу, воздействовать на целостность клеточных мембран, стимулировать синтез глутатиона и воздействовать на депозиты ?-амилоида [4, 32].
В клинических исследованиях доказано влияние на когнитивную функцию у пациентов после инсульта при 2-летнем наблюдении за 347 пожилыми пациентами в двух группах (67,2±11,3), перенесшими инсульт и имевшими когнитивные нарушения (цитиколин в дозе 1000 мг/сут. peros в течение 12 месяцев против группы контроля). По результатам оценочных тестов (Батарея лобной дисфункции, внимание, исполнительные (регуляторные) функции, ориентацию во времени и др.) отмечено значимое улучшение когнитивных функций (речь, память, ориентирование в пространстве и во времени, скорость моторики) [5]. При когнитивных нарушениях, развивающихся вследствие сосудистых нарушений, отмечены аналогичные результаты [17].
В обзоре наиболее значительных опубликованных клинических исследований по снижению познавательной способности в среднесрочной и долгосрочной перспективе на сосудистых когнитивных нарушениях и при БА с использованием цитиколина приводятся данные, подтверждающие благотворное влияние лекарственного средства, но указывается на методологическую гетерогенность этих исследований, что затрудняет заключительные выводы [24]. В небольшом российском исследовании у 54 пациентов с СД с диабетической энцефалопатией у 77,8% пациентов лечение цитиколином приводило к улучшению состояния, подтвержденному результатами выполнения нейропсихологических тестов [1]. В заключение авторы отмечают, что цитиколин положительно влияет на мозговую гемодинамику посредством повышения средней скорости кровотока по интракраниальным артериям каротидного и вертебробазилярного бассейнов и уменьшает асимметрию кровотока; комплексно улучшает мозговое кровообращение, функцию эндотелия сосудов головного мозга и когнитивные функции у больных с диабетической энцефалопатией. Для окончательного понимания эффективности цитиколина при когнитивных нарушениях у пациентов с СД необходимо проведение исследования со стандартизованными диагностическими критериями включения и стандартизованной нейропсихологической оценкой и использование современных методов нейровизуализации (функциональная МРТ).
В перечне потенциальных воздействий может обсуждаться использование различных лекарственных средств с антиоксидантным влиянием. Поскольку доказана роль дефицита витамина В12, альтернативным вмешательством может быть назначение этого лекарственного средства и других витаминов с антиоксидантным и нейротропным влиянием. В небольшом одноцентровом рандомизированном исследовании VITACOG (271 пациент с легкими когнитивными нарушениями в возрасте 70 лет и старше) было показано, что применение высоких доз фолиевой кислоты и витаминов В6 и В12 приводит к позитивным изменениям по результатам оценки объема и функций мозга [18].
Для пациентов с БА в комплексную терапию включают холиномиметик центрального действия – холина альфосцерат – антихолинэстеразный препарат, стимулирующий нервно-мышечную и центральную холинергическую передачу ипидакрина, селективный обратимый длительно действующий ингибитор ацетилхолинэстеразы галантамин, антагонист NMDA-рецепторов, регулирует ионный транспорт (блокирует калиевые каналы) мемантин.
В большинстве случаев СД 2-го типа когнитивные нарушения развиваются у пациентов в возрасте старше 50 лет, что ассоциировано с возрастным снижением секреции половых гормонов. С этой точки зрения представляют интерес доказательства связи дефицита эстрогенов и прогестагенов с когнитивными нарушениями. Доказано, что в эксперименте прогестерон обеспечивает нейропротективный эффект, способствуя выживанию нейронов после травмы и дегенеративных заболеваний, и миелинизирующий эффект с улучшением формирования миелиновых оболочек [49]. Данная публикация не посвящена детальному анализу преимуществ различных вариантов заместительной менопаузальной терапии, однако комбинация эстрадиола и микронизированного прогестерона улучшает когнитивные функции (настроение, оперативная память и долгосрочная память).
В заключение следует отметить, что рост заболеваемости СД и увеличение продолжительности жизни пациентов с этой патологией диктуют необходимость актуализации изучения когнитивных расстройств, включая механизм их развития, внедрение нейропсихологических методов исследования – простых скрининговых шкал – в клиническую практику и разработку доказательных лечебных подходов для этой категории пациентов.
Л И Т Е Р А Т У Р А
1. Мельник Т.М. // Архив клинической и экспериментальной медицины. – 2011. – №2. – С.182–185.
2. Чугунов П.А., Семенова И.В. // Сахарный диабет. – 2008. – №1 (38). – С.61–68.
3. Шмырев В.И., Остроумова О.Д., Боброва Т.А. // Русс. мед. журн. – 2003. – №4. – С.216–220.
4. Álvarez-Sabín J., Román G.C. // Brain Sci. – 2013. – Vol.3, N3. – P.1395–1414.
5. Alvarez-Sabín J., Santamarina E., Maisterra O., et al. // Int. J. Mol. Sci. – 2016. – Vol.17, N3. – P.390.
6. Aroda V.R., Edelstein S.L., Goldberg R.B., et al. // JCEM. – 2016. – Vol.101, N4. – P.1754–1761.
7. Аrboleda G., Huang T.J., Waters C., et al. // Eur. J. Neurosci. – 2007. – Vol.25, N10. – P.3030–3038.
8. Barrett E.J., Liu Z., Khamaisi M., et al. // JCEM. – 2017. – Vol.102, N12. – P.4343–4410.
9. Bhateja D.K., Dhull D.K., Gill A., et al. // Eur. J. Pharmacology. – 2011. – Vol.674, N1. – P.33–43.
10. Biessels G.J., Staekenborg S., Brunner E., et al. // Lancet Neurol. – 2006. – Vol.5. – P. 64–74.
11. Bosch J., et al. // American Heart Association Scientific Sessions. – 2016. – Vol.12–16.
12. Bree A.J., Puente E.C., Daphna-Iken D., Fisher S.J. // Am. J. Physiol. Endocrinol. Metab. – 2009. – Vol.297, N1. – E194–E201.
13. Castellani R.J., Harris P.L., Sayre L.M., et al. // Free Radic. Biol. Med. – 2001. – Vol.31, N2. – P.175–180.
14. Cederholm T., Salem N.Jr, Palmblad J. // Adv. Nutr. – 2013. – Vol.4, N6. – P.672–676.
15. Chen J., Li S., Sun W., Li J. // PLoS One. – 2015. – Vol.10, N4. – e0123864.249.
16. Cheignon С., Tomas M., Bonnefont-Rousselot D., et al. // Redox. Biol. – 2018. – Vol.14. – P.450–464.
17. Cotroneo A.M., Castagna A., Putignano S., et al. // Clin. Interv. Aging. – 2013. – Vol.8. – P.131–137.
18. de Jager C.A., Oulhaj A., Jacoby R., et al. // Int. J. Geriatr. Psychiatry. – 2012. – Vol.27, N6. – P.592–600.
19. de la Monte S.M., Tong M., VanAnhNguyen, et al. // J. Alzheimers Dis. – 2010. – Vol.21, N3. – P.967–984.
20. Derev’yannykh E.A., Bel’skaya G.N., Knoll E.A., et al. // Neurosci. Behav. Physiol. – 2008. – Vol.38. – P.873–875.
21. Elmlinger M.W., Kriebel M., Ziegler D. // Neuromolecular. Med. – 2011. – Vol.13. – P.266–274.
22. Feinkohl I., Price J.F., Strachan M.W.J., Frier B.M. // Res. Ther. – 2015. – Vol.7, N1. – P.46.
23. García-Casares N., García-Arnés J.A., Gómez-Huelgas R., Valdivielso-Felices P., García-Arias C., González-Santos P. // Rev. Neurol. – 2014. – Vol.59, N11. – P.517–524.
24. García-Cobos R., Frank-García A., Gutiérrez-Fernández M., et al. // J. Neurological Sciences 2010. – Vol.299, N1–2. – P.188–192.
25. Gispen W.H., Biessels G.J. // Eur. J. Pharmacol. – 2000. – Vol.223. – P.656–658.
26. Ghasemi R., Haeri A., Dargahi L., et al. // Mol. Neurobiol. – 2013. – Vol.47. – P.145–147.
27. Gorelick P.B. // Ann. NY Acad. Sci. – 2010. – Vol.1207. – P.155–162.
28. Gray S.M., Meijer R.I., Barrett E.J. // Diabetes. – 2014. – Vol.63, N12. – P.3992–3997.
29. Guekht A., Skoog I., Edmundson S., et al. // Stroke. – 2017. – Vol.48. – P.1262–1270.
30. Hebert L.E., Weuve J., Scherr P.A., Evans D.A. // Neurology. – 2013. – Vol.80, N19. – P.1778–1783.
31. Holscher C. // Sheng Li Xue Bao. – 2014. – Vol.66, N5. – P.497–510.
32. Hurtado O., Lizasoain I., Moro M.Á. // Stroke. – 2011. – Vol.42 (1 Suppl). – P.33–35.
33. Li J., Deng J., Sheng W., Zuo Z. // Pharmacol. Biochem. Behav. – 2012. – Vol.101, N4. – P.564–574.
34. Lin C.H., Sheu W.H. // J. Intern. Med. – 2013. – Vol.273, N1. – P.102–110.
35. Lonn E.M., Bosch J., López-Jaramillo P., et al. // NEJM. – 2016. – Vol.374, N21. – P.2009–2020.
36. Lundbæk K. // Acta Neurologic. Scandinavica. – 1963. – doi:10.1111/j.1600-0404.1963.tb01844
37. Machicao F., Muresanu D.F., Hundsberger H., et al. // J. Neurol. Sci. – 2012. – Vol.322. – P.222–227.
38. Manschot S.M., Brands A.M., van der Grond J., et al. // Diabetes. – 2006. – Vol.55. – P.1106–1113.
39. McEwen B.S., Reagan L.P. // Eur. J. Pharmacol. – 2004. – Vol.490. – P.13–24.
40. McGuinness B., Craig D., Bullock R., Malouf R., Passmore P. // Cochrane Database Syst. Rev. – 2014. – Vol.7. – CD007514.
41. Mittal K., Mani R.J., Katare D.P. // Sci. Rep. – 2016. – Vol.6. – P.25589. doi:10.1038/srep25589
42. Musen G., Lyoo I.K., Sparks C.R., et al. // Diabetes. – 2006. – Vol.55, N2. – P.326–333.
43. Nigel Irwin, Peter R. Flatt. // World J. Diabetes. – 2015. – Vol.6, N15. – P.1285–1295.
44. Patrick A.W., Campbell I.W. // Diabet Med. – 1990. – Vol.7. – P.349–354.
45. Peters R., Beckett N., Forette F., et al. // Lancet Neurol. – 2008. – Vol.7, N8. – P.683–689.
46. Reske-Nielsen E., Kroner Z. // Altern. Med. Rev. –2009. – Vol.14, N4. – P.373–379.
47. Saedi E., Gheini M.R., Faiz F., Arami M.A. // World J. Diabetes. – 2016. – Vol.7, N17. – P.412–422.
48. Schilling T.M., Ferreira de Sa D.S., Westerhausen R., Strelzyk F., Larra M.F., Hallschmid M., et al. // Hum. Brain Mapp. – 2014. – Vol.35, N5. – P.1944–1956.
49. Schumacher M., Hussain R., Gago N., et al. // Front Neurosci. – 2012. – Vol.6. – P.10.
50. Swift D.L., Johannsen N.M., Myers V.H., et al. // PLoS One. – 2012. – Vol.7, N8. – e42785. doi:10.1371/journal.pone.0042785
51. Toba J., Nikkuni M., Ishizeki M., et al. // Biochem. Biophys. Res. Commun. – 2016. – Vol.473, N4. – P.1039–1044.
52. Tong M., de la Monte S.M. // J. Alzheimers Dis. – 2009. – Vol.16, N4. – P.705–714.
53. Sherwin B.B., Grigorova M. // Fertil Steril. – 2011. – Vol.96. – P.399–403.
54. van Harten B., de Leeuw F.E., Weinstein H.C., et al. // Diabetes Care. – 2006. – Vol.29, N11. – P.2539–2548.
55. Wessels A.M., Simsek S., Remijnse P.L. // Diabetologia. – 2006. – Vol.49, N10. – P.2474–2480.
56. Whitmer R.A. // Curr. Alzheimer Res. – 2007. – Vol.4, N2. – P.117–122.
57. Wilkosc M., Markowska A., Zajac-Lamparska L., et al. // Front Neural Circuits. – 2016. – Vol.10. – P.39.
58. Xu W.L., von Strauss E., Qiu C.X., et al. // Diabetologia. – 2009. – Vol.52, N6. – P.1031–1039.
59. Zou C., Shi Y., Ohli J., et al. // Acta Neuropathol. – 2016. – Vol.131, N2. – P.235–246.
Медицинские новости. – 2018. – №10. – С. 13-20.
Внимание! Статья адресована врачам-специалистам. Перепечатка данной статьи или её фрагментов в Интернете без гиперссылки на первоисточник рассматривается как нарушение авторских прав.